Data preparation
We just use the dataset which are from previous step.
library(tidymass)
#> Registered S3 method overwritten by 'Hmisc':
#> method from
#> vcov.default fit.models
#> ── Attaching packages ──────────────────────────────────────── tidymass 1.0.9 ──
#> ✔ massdataset 1.0.34 ✔ metid 1.2.32
#> ✔ massprocesser 1.0.10 ✔ masstools 1.0.13
#> ✔ masscleaner 1.0.12 ✔ dplyr 1.1.4
#> ✔ massqc 1.0.7 ✔ ggplot2 3.5.1
#> ✔ massstat 1.0.6 ✔ magrittr 2.0.3
#> ✔ metpath 1.0.8
load("data_cleaning/POS/object_pos2")
load("data_cleaning/NEG/object_neg2")
Add MS2 spectra data to mass_dataset class
Download the MS2 data here.
Uncompress it.
Positive mode
object_pos2 <-
mutate_ms2(
object = object_pos2,
column = "rp",
polarity = "positive",
ms1.ms2.match.mz.tol = 15,
ms1.ms2.match.rt.tol = 30,
path = "mgf_ms2_data/POS"
)
#> Reading mgf data...
#> Reading mgf data...
#> Reading mgf data...
#> Reading mgf data...
#> 1042 out of 5101 variable have MS2 spectra.
#> Selecting the most intense MS2 spectrum for each peak...
object_pos2
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 5101 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 5101 x 6 data.frame]
#> 5101 variables:M70T53_POS M70T527_POS M71T775_POS ... M836T610_POS M836T759_POS
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 6 x 2 data.frame]
#> 6.ms2_data:[ 1042 variables x 951 MS2 spectra]
#> --------------------
#> Processing information
#> 9 processings in total
#> Latest 3 processings show
#> normalize_data ----------
#> Package Function.used Time
#> 1 masscleaner normalize_data() 2023-08-31 14:10:01
#> integrate_data ----------
#> Package Function.used Time
#> 1 masscleaner integrate_data() 2023-08-31 14:10:03
#> mutate_ms2 ----------
#> Package Function.used Time
#> 1 massdataset mutate_ms2() 2024-09-10 10:51:52
extract_ms2_data(object_pos2)
#> $`QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf`
#> --------------------
#> column: rp
#> polarity: positive
#> mz_tol: 15
#> rt_tol (second): 30
#> --------------------
#> 1042 variables:
#> M71T775_POS M72T53_POS M83T50_POS M84T57_POS M85T54_POS...
#> 951 MS2 spectra.
#> mz70.981170654297rt775.4286 mz72.081642150879rt53.6528862 mz82.945625305176rt49.238013 mz84.045127868652rt59.6895132 mz85.029016959043rt53.0835648...
Negative mode
object_neg2 <-
mutate_ms2(
object = object_neg2,
column = "rp",
polarity = "negative",
ms1.ms2.match.mz.tol = 15,
ms1.ms2.match.rt.tol = 30,
path = "mgf_ms2_data/NEG"
)
#> Reading mgf data...
#> Reading mgf data...
#> Reading mgf data...
#> Reading mgf data...
#> 1092 out of 4104 variable have MS2 spectra.
#> Selecting the most intense MS2 spectrum for each peak...
object_neg2
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 4104 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 4104 x 6 data.frame]
#> 4104 variables:M70T712_NEG M70T587_NEG M71T587_NEG ... M884T57_NEG M899T56_NEG
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 6 x 2 data.frame]
#> 6.ms2_data:[ 1092 variables x 988 MS2 spectra]
#> --------------------
#> Processing information
#> 9 processings in total
#> Latest 3 processings show
#> normalize_data ----------
#> Package Function.used Time
#> 1 masscleaner normalize_data() 2023-08-31 14:10:13
#> integrate_data ----------
#> Package Function.used Time
#> 1 masscleaner integrate_data() 2023-08-31 14:10:14
#> mutate_ms2 ----------
#> Package Function.used Time
#> 1 massdataset mutate_ms2() 2024-09-10 10:52:05
extract_ms2_data(object_neg2)
#> $`QEP_SGA_QC_neg_ms2_ce25_01.mgf;QEP_SGA_QC_neg_ms2_ce25_02.mgf;QEP_SGA_QC_neg_ms2_ce50_01.mgf;QEP_SGA_QC_neg_ms2_ce50_02.mgf`
#> --------------------
#> column: rp
#> polarity: negative
#> mz_tol: 15
#> rt_tol (second): 30
#> --------------------
#> 1092 variables:
#> M71T51_NEG M73T74_NEG M75T52_NEG M80T299_NEG M80T232_NEG...
#> 988 MS2 spectra.
#> mz71.012359619141rt52.3270968 mz73.02799987793rt74.779476 mz75.007308959961rt24.1557228 mz79.955728954783rt301.268466 mz79.955834350356rt235.127328...
Metabolite annotation
Metabolite annotation is based on the metid package.
Download database
We need to download MS2 database from metid website.
Here we download the Michael Snyder RPLC databases, Orbitrap database and MoNA database. And place them in a new folder named as metabolite_annotation.
Positive mode
Annotate features using snyder_database_rplc0.0.3.
load("metabolite_annotation/snyder_database_rplc0.0.3.rda")
snyder_database_rplc0.0.3
#> -----------Base information------------
#> Version:0.0.2
#> Source:MS
#> Link:http://snyderlab.stanford.edu/
#> Creater:Xiaotao Shen(shenxt1990@163.com)
#> With RT information
#> -----------Spectral information------------
#> 14 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 917 metabolites in total.
#> 356 metabolites with spectra in positive mode.
#> 534 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:2
#> NCE25; NCE50
#> Collision energy in negative mode:
#> Total number:2
#> NCE25; NCE50
object_pos2 <-
annotate_metabolites(
object = object_pos2,
database = snyder_database_rplc0.0.3,
based_on = c("ms1", "rt", "ms2"),
polarity = "positive",
column = "rp",
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 2%
|
|== | 3%
|
|== | 4%
|
|=== | 4%
|
|=== | 5%
|
|==== | 5%
|
|==== | 6%
|
|===== | 7%
|
|====== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======= | 11%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 17%
|
|============ | 18%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|============== | 21%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================= | 24%
|
|================= | 25%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 26%
|
|=================== | 27%
|
|=================== | 28%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 30%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 40%
|
|============================ | 41%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================= | 48%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 49%
|
|=================================== | 50%
|
|=================================== | 51%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 59%
|
|========================================== | 60%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 70%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 72%
|
|=================================================== | 73%
|
|=================================================== | 74%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 82%
|
|========================================================== | 83%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 89%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 93%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 100%
#> All done.
Annotate features using orbitrap_database0.0.3.
load("metabolite_annotation/orbitrap_database0.0.3.rda")
orbitrap_database0.0.3
#> -----------Base information------------
#> Version:0.0.1
#> Source:NIST
#> Link:https://www.nist.gov/
#> Creater:Xiaotao Shen(shenxt1990@163.com)
#> Without RT informtaion
#> -----------Spectral information------------
#> 8 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula
#> 8360 metabolites in total.
#> 7103 metabolites with spectra in positive mode.
#> 3311 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:12
#> 10; 15; 45; 55; 5; 20; 30; 35; 40; 25
#> Collision energy in negative mode:
#> Total number:12
#> 10; 25; 5; 15; 20; 30; 50; 35; 40; 45
object_pos2 <-
annotate_metabolites(
object = object_pos2,
ms1.match.ppm = 15,
polarity = "positive",
column = "rp",
database = orbitrap_database0.0.3,
based_on = c("ms1", "ms2"),
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
| | 1%
|
|= | 1%
|
|= | 2%
|
|== | 2%
|
|== | 3%
|
|== | 4%
|
|=== | 4%
|
|=== | 5%
|
|==== | 5%
|
|==== | 6%
|
|===== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 8%
|
|====== | 9%
|
|======= | 9%
|
|======= | 10%
|
|======= | 11%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========= | 14%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 16%
|
|============ | 17%
|
|============ | 18%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|============== | 21%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================ | 24%
|
|================= | 24%
|
|================= | 25%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 26%
|
|=================== | 27%
|
|=================== | 28%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 29%
|
|===================== | 30%
|
|===================== | 31%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================= | 34%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 36%
|
|========================== | 37%
|
|========================== | 38%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 39%
|
|============================ | 40%
|
|============================ | 41%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|============================== | 44%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 46%
|
|================================= | 47%
|
|================================= | 48%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 49%
|
|=================================== | 50%
|
|=================================== | 51%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 52%
|
|===================================== | 53%
|
|===================================== | 54%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 59%
|
|========================================== | 60%
|
|========================================== | 61%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 62%
|
|============================================ | 63%
|
|============================================ | 64%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 69%
|
|================================================= | 70%
|
|================================================= | 71%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 72%
|
|=================================================== | 73%
|
|=================================================== | 74%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 82%
|
|========================================================== | 83%
|
|========================================================== | 84%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 89%
|
|=============================================================== | 90%
|
|=============================================================== | 91%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 92%
|
|================================================================= | 93%
|
|================================================================= | 94%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 99%
|
|======================================================================| 100%
#> All done.
Annotate features using mona_database0.0.3.
load("metabolite_annotation/mona_database0.0.3.rda")
mona_database0.0.3
#> -----------Base information------------
#> Version:0.0.1
#> Source:MoNA
#> Link:http://mona.fiehnlab.ucdavis.edu/
#> Creater:Xiaotao Shen(shenxt1990@163.com)
#> Without RT informtaion
#> -----------Spectral information------------
#> 25 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; CE; Chebi.ID (top10)
#> 19537 metabolites in total.
#> 9307 metabolites with spectra in positive mode.
#> 10243 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:284
#> Ramp 21.1-31.6; Ramp 20.8-31.3; 20; 30; 40; 50; 10; Ramp 21.8-32.7; Ramp 18.5-27.8; Ramp 20.5-30.7
#> Collision energy in negative mode:
#> Total number:217
#> 10; 20; 30; 40; 50; 35 % (nominal); 15 % (nominal); 30 % (nominal); 45 % (nominal); 60 % (nominal)
object_pos2 <-
annotate_metabolites(
object = object_pos2,
ms1.match.ppm = 15,
polarity = "positive",
column = "rp",
database = mona_database0.0.3,
based_on = c("ms1", "ms2"),
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
| | 1%
|
|= | 1%
|
|= | 2%
|
|== | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 5%
|
|==== | 6%
|
|===== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 8%
|
|====== | 9%
|
|======= | 9%
|
|======= | 10%
|
|======= | 11%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========= | 14%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 16%
|
|============ | 17%
|
|============ | 18%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|============== | 21%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================= | 24%
|
|================= | 25%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|=================== | 28%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 29%
|
|===================== | 30%
|
|===================== | 31%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================= | 34%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 36%
|
|========================== | 37%
|
|========================== | 38%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 39%
|
|============================ | 40%
|
|============================ | 41%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|============================== | 44%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================= | 48%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 49%
|
|=================================== | 50%
|
|=================================== | 51%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 59%
|
|========================================== | 60%
|
|========================================== | 61%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 62%
|
|============================================ | 63%
|
|============================================ | 64%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 69%
|
|================================================= | 70%
|
|================================================= | 71%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 82%
|
|========================================================== | 83%
|
|========================================================== | 84%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 89%
|
|=============================================================== | 90%
|
|=============================================================== | 91%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 92%
|
|================================================================= | 93%
|
|================================================================= | 94%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 99%
|
|======================================================================| 100%
#> All done.
Negative mode
Annotate features using snyder_database_rplc0.0.3.
object_neg2 <-
annotate_metabolites(
object = object_neg2,
ms1.match.ppm = 15,
rt.match.tol = 30,
polarity = "negative",
column = "rp",
database = snyder_database_rplc0.0.3,
based_on = c("ms1", "rt", "ms2"),
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
|= | 1%
|
|== | 2%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 6%
|
|===== | 7%
|
|====== | 8%
|
|====== | 9%
|
|======= | 9%
|
|======= | 10%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========== | 14%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 29%
|
|===================== | 30%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 40%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 70%
|
|================================================= | 71%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 90%
|
|=============================================================== | 91%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 93%
|
|================================================================= | 94%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 100%
#> All done.
Annotate features using orbitrap_database0.0.3.
object_neg2 <-
annotate_metabolites(
object = object_neg2,
ms1.match.ppm = 15,
polarity = "negative",
column = "rp",
database = orbitrap_database0.0.3,
based_on = c("ms1", "ms2"),
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
| | 1%
|
|= | 1%
|
|= | 2%
|
|== | 2%
|
|== | 3%
|
|=== | 4%
|
|=== | 5%
|
|==== | 5%
|
|==== | 6%
|
|===== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 8%
|
|====== | 9%
|
|======= | 9%
|
|======= | 10%
|
|======= | 11%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 16%
|
|============ | 17%
|
|============ | 18%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|============== | 21%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================ | 24%
|
|================= | 24%
|
|================= | 25%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|=================== | 28%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 29%
|
|===================== | 30%
|
|===================== | 31%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================= | 34%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 36%
|
|========================== | 37%
|
|========================== | 38%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 39%
|
|============================ | 40%
|
|============================ | 41%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 46%
|
|================================= | 47%
|
|================================= | 48%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 49%
|
|=================================== | 50%
|
|=================================== | 51%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 52%
|
|===================================== | 53%
|
|===================================== | 54%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 59%
|
|========================================== | 60%
|
|========================================== | 61%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 62%
|
|============================================ | 63%
|
|============================================ | 64%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 69%
|
|================================================= | 70%
|
|================================================= | 71%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 82%
|
|========================================================== | 83%
|
|========================================================== | 84%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 89%
|
|=============================================================== | 90%
|
|=============================================================== | 91%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 92%
|
|================================================================= | 93%
|
|================================================================= | 94%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 99%
|
|======================================================================| 100%
#> All done.
Annotate features using mona_database0.0.3.
object_neg2 <-
annotate_metabolites(
object = object_neg2,
ms1.match.ppm = 15,
polarity = "negative",
column = "rp",
database = mona_database0.0.3,
based_on = c("ms1", "ms2"),
adduct.table = NULL
)
#> No adduct table is provided. Use the default adduct table.
#> Checking parameters...
#> All parameters are correct.
#>
#> Use all CE values.
#>
|
| | 0%
|
| | 1%
|
|= | 1%
|
|= | 2%
|
|== | 2%
|
|== | 3%
|
|== | 4%
|
|=== | 4%
|
|=== | 5%
|
|==== | 5%
|
|==== | 6%
|
|===== | 6%
|
|===== | 7%
|
|===== | 8%
|
|====== | 8%
|
|====== | 9%
|
|======= | 9%
|
|======= | 10%
|
|======= | 11%
|
|======== | 11%
|
|======== | 12%
|
|========= | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 15%
|
|=========== | 16%
|
|============ | 16%
|
|============ | 17%
|
|============ | 18%
|
|============= | 18%
|
|============= | 19%
|
|============== | 19%
|
|============== | 20%
|
|============== | 21%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 22%
|
|================ | 23%
|
|================= | 24%
|
|================= | 25%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|=================== | 28%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 29%
|
|===================== | 30%
|
|===================== | 31%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 32%
|
|======================= | 33%
|
|======================= | 34%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|========================== | 38%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 39%
|
|============================ | 40%
|
|============================ | 41%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 42%
|
|============================== | 43%
|
|============================== | 44%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 46%
|
|================================= | 47%
|
|================================= | 48%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 49%
|
|=================================== | 50%
|
|=================================== | 51%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 52%
|
|===================================== | 53%
|
|===================================== | 54%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 56%
|
|======================================== | 57%
|
|======================================== | 58%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 59%
|
|========================================== | 60%
|
|========================================== | 61%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================= | 65%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 66%
|
|=============================================== | 67%
|
|=============================================== | 68%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 69%
|
|================================================= | 70%
|
|================================================= | 71%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|====================================================== | 78%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 79%
|
|======================================================== | 80%
|
|======================================================== | 81%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 82%
|
|========================================================== | 83%
|
|========================================================== | 84%
|
|=========================================================== | 84%
|
|=========================================================== | 85%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================= | 88%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 89%
|
|=============================================================== | 90%
|
|=============================================================== | 91%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 92%
|
|================================================================= | 93%
|
|================================================================= | 94%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 96%
|
|==================================================================== | 97%
|
|==================================================================== | 98%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 99%
|
|======================================================================| 100%
#> All done.
Annotation result
The annotation result will assign into mass_dataset class as the annotation_table slot.
head(extract_annotation_table(object = object_pos2))
#> variable_id
#> 1 M100T160_POS
#> 2 M103T100_POS
#> 3 M103T100_POS
#> 4 M104T51_POS
#> 5 M113T187_POS
#> 6 M113T81_POS
#> ms2_files_id
#> 1 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> 2 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> 3 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> 4 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> 5 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> 6 QEP_SGA_QC_posi_ms2_ce25_01.mgf;QEP_SGA_QC_posi_ms2_ce25_02.mgf;QEP_SGA_QC_posi_ms2_ce50_01.mgf;QEP_SGA_QC_posi_ms2_ce50_02.mgf
#> ms2_spectrum_id Compound.name CAS.ID HMDB.ID
#> 1 mz100.076248168945rt158.377638 N-Methyl-2-pyrrolidone 872-50-4 <NA>
#> 2 mz103.054814801682rt96.92601 Phenylacetaldehyde 122-78-1 HMDB06236
#> 3 mz103.054814801682rt96.92601 3-Amino-2-oxazolidinone 80-65-9 <NA>
#> 4 mz104.107467651367rt49.510314 5-Amino-1-pentanol 2508-29-4 <NA>
#> 5 mz113.060150146484rt188.406384 1,4-Cyclohexanedione <NA> <NA>
#> 6 mz113.035087585449rt77.20827 URACIL <NA> <NA>
#> KEGG.ID Lab.ID Adduct mz.error mz.match.score RT.error
#> 1 C11118 MONA_11509 (M+H)+ 1.335652 0.9960435 NA
#> 2 C00601 NO07389 (M+H-H2O)+ 1.537004 0.9947640 NA
#> 3 <NA> NO07231 (M+H)+ 11.537004 0.7439487 NA
#> 4 <NA> NO07238 (M+H)+ 1.169128 0.9969671 NA
#> 5 <NA> MONA_14519 (M+H)+ 1.051626 0.9975454 NA
#> 6 <NA> MONA_18148 (M+H)+ 1.275544 0.9963909 NA
#> RT.match.score CE SS Total.score Database Level
#> 1 NA 35 (nominal) 0.6871252 0.8029696 MoNA_0.0.1 2
#> 2 NA 10 0.5748835 0.7323387 NIST_0.0.1 2
#> 3 NA 20 0.5020256 0.5927468 NIST_0.0.1 2
#> 4 NA 5 0.5971697 0.7470937 NIST_0.0.1 2
#> 5 NA HCD (NCE 20-30-40%) 0.5401414 0.7116679 MoNA_0.0.1 2
#> 6 NA 10 0.6889885 0.8042644 MoNA_0.0.1 2
variable_info_pos <-
extract_variable_info(object = object_pos2)
head(variable_info_pos)
#> variable_id mz rt na_freq na_freq.1 na_freq.2 Compound.name
#> 1 M70T53_POS 70.06596 52.78542 0.00000000 0.14545455 0.00000000 <NA>
#> 2 M70T527_POS 70.36113 526.76657 0.02564103 0.18181818 0.30000000 <NA>
#> 3 M71T775_POS 70.98125 775.44867 0.00000000 0.00000000 0.00000000 <NA>
#> 4 M71T669_POS 70.98125 668.52844 0.00000000 0.02727273 0.01818182 <NA>
#> 5 M71T715_POS 70.98125 714.74066 0.05128205 0.12727273 0.02727273 <NA>
#> 6 M71T54_POS 71.04999 54.45641 0.15384615 0.99090909 0.05454545 <NA>
#> CAS.ID HMDB.ID KEGG.ID Lab.ID Adduct mz.error mz.match.score RT.error
#> 1 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> 2 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> 3 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> 4 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> 5 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> 6 <NA> <NA> <NA> <NA> <NA> NA NA NA
#> RT.match.score CE SS Total.score Database Level
#> 1 NA <NA> NA NA <NA> NA
#> 2 NA <NA> NA NA <NA> NA
#> 3 NA <NA> NA NA <NA> NA
#> 4 NA <NA> NA NA <NA> NA
#> 5 NA <NA> NA NA <NA> NA
#> 6 NA <NA> NA NA <NA> NA
table(variable_info_pos$Level)
#>
#> 1 2
#> 23 183
table(variable_info_pos$Database)
#>
#> MoNA_0.0.1 MS_0.0.2 NIST_0.0.1
#> 78 23 105
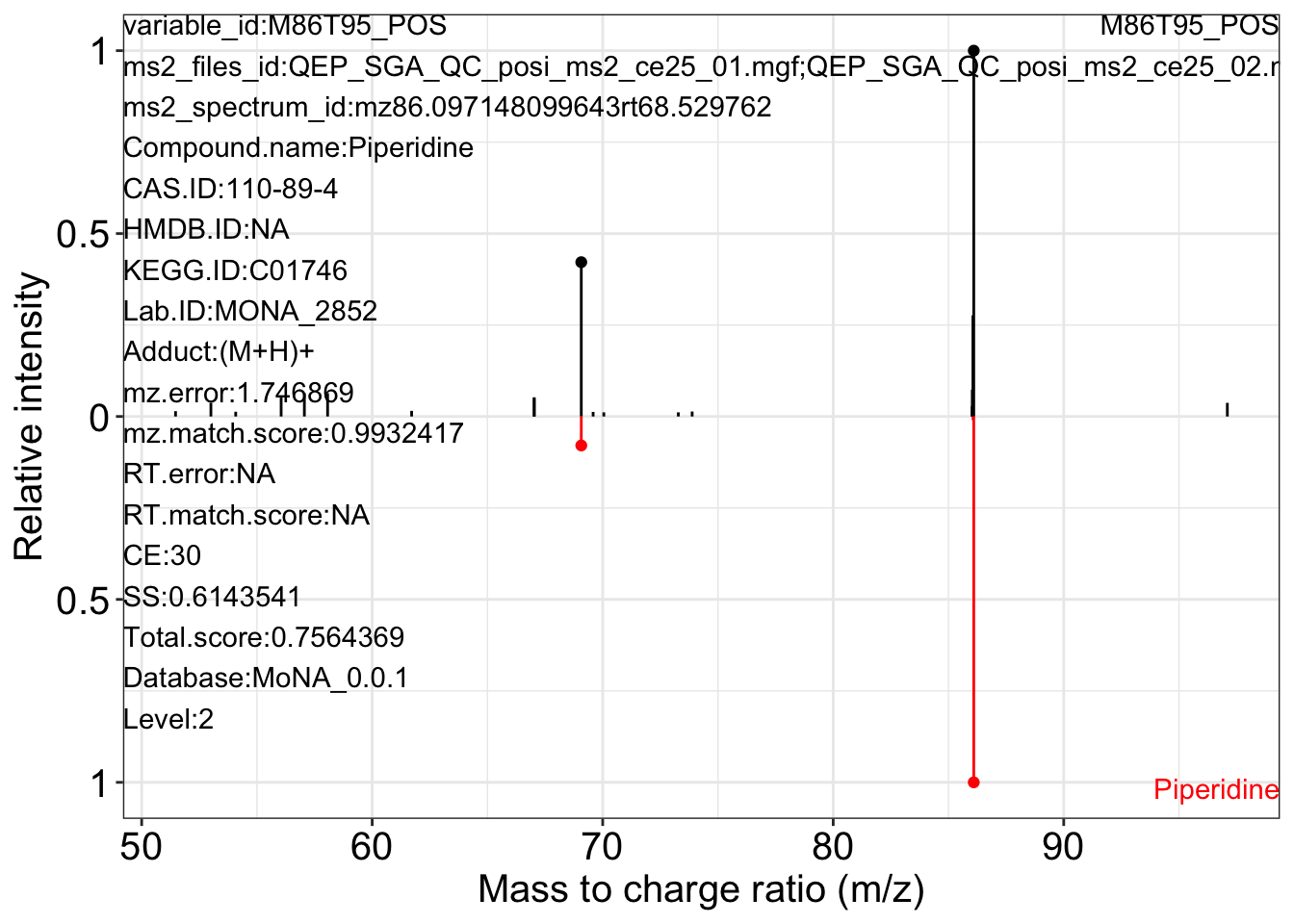
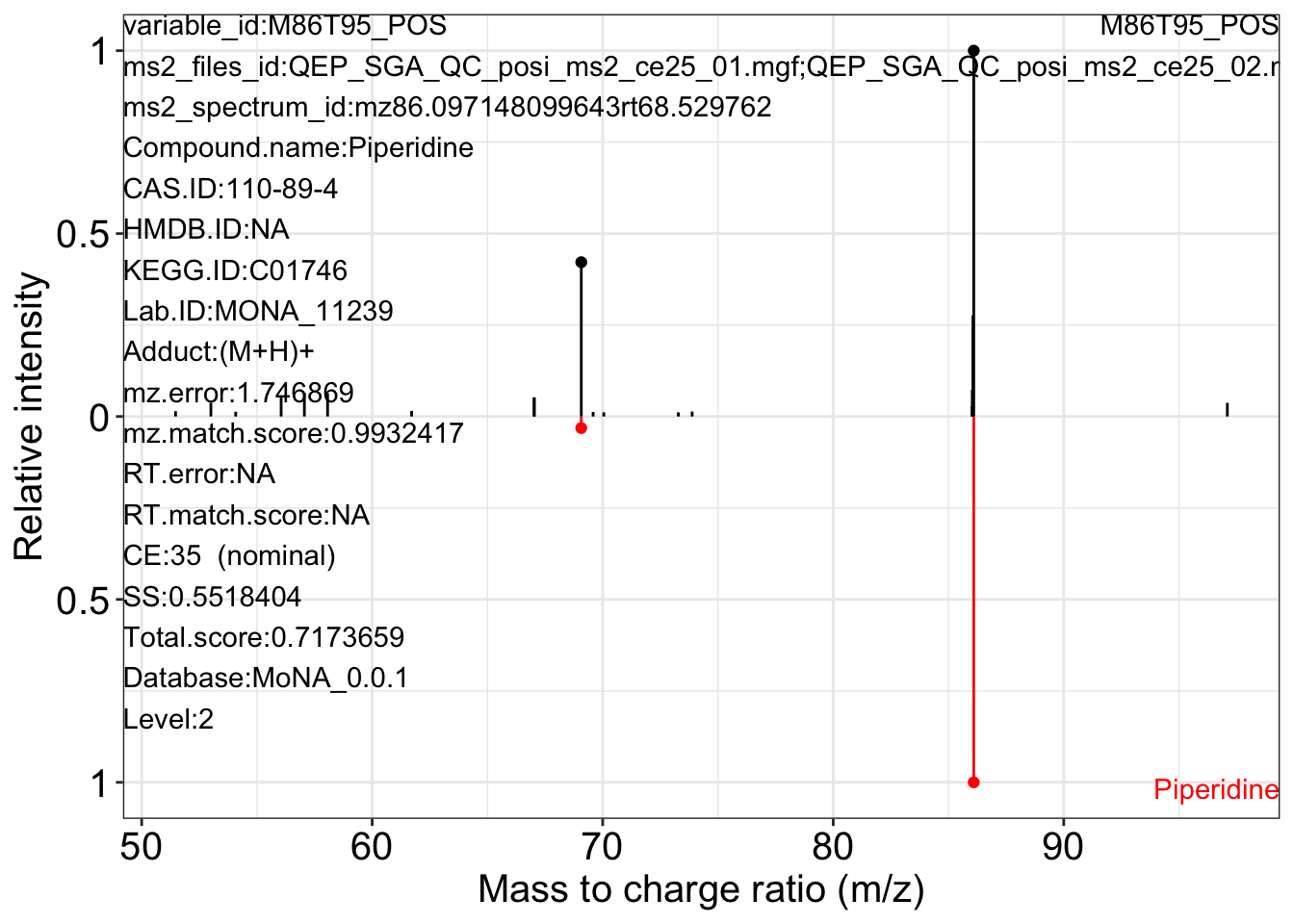
Use the ms2_plot_mass_dataset() function to get the MS2 matching plot.
ms2_plot_mass_dataset(object = object_pos2,
variable_id = "M86T95_POS",
database = mona_database0.0.3)
#> $M86T95_POS_1
#>
#> $M86T95_POS_2
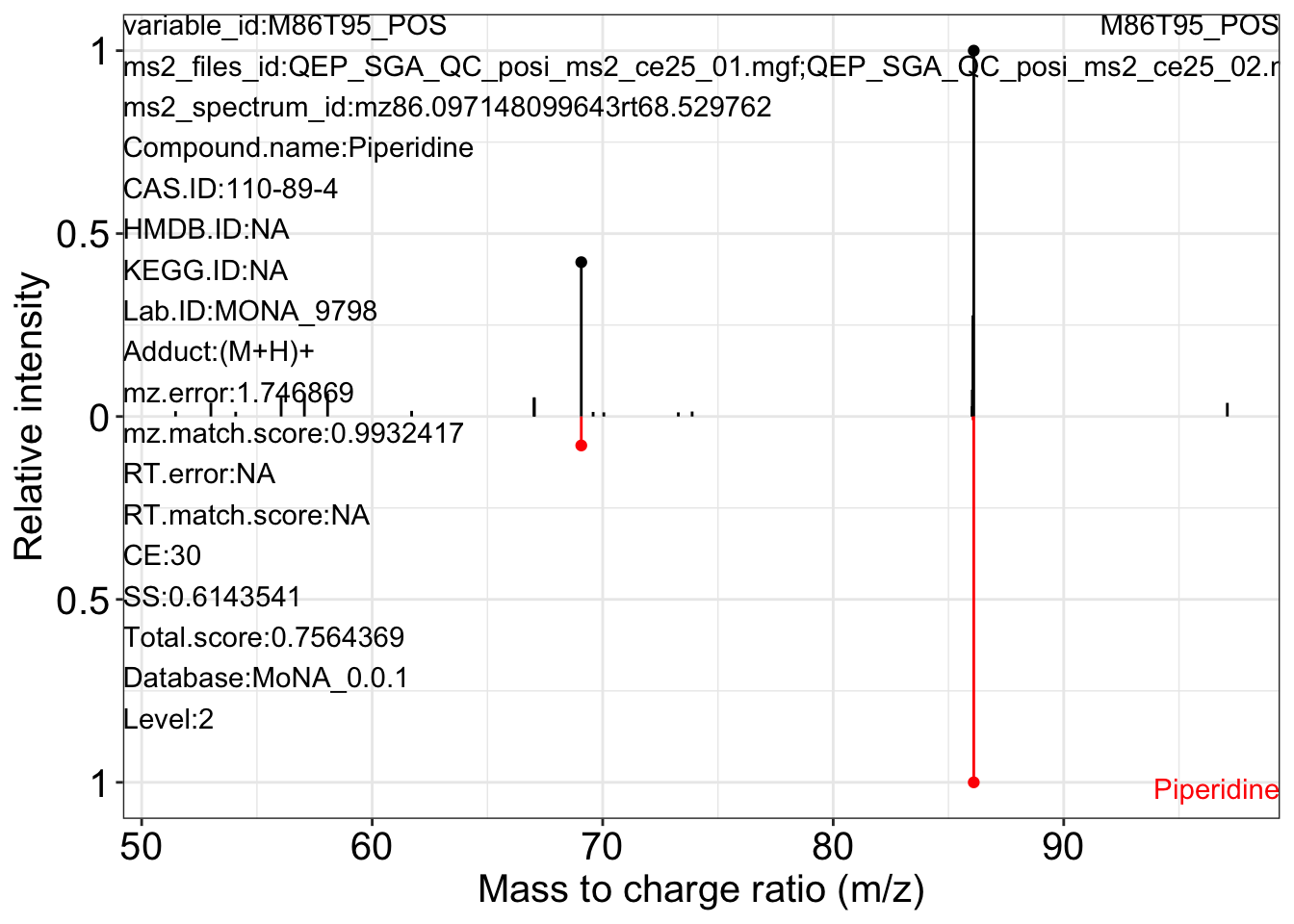
#>
#> $M86T95_POS_3
ms2_plot_mass_dataset(object = object_pos2,
variable_id = "M86T95_POS",
database = mona_database0.0.3,
interactive_plot = TRUE)
#> $M86T95_POS_1
#>
#> $M86T95_POS_2
#>
#> $M86T95_POS_3
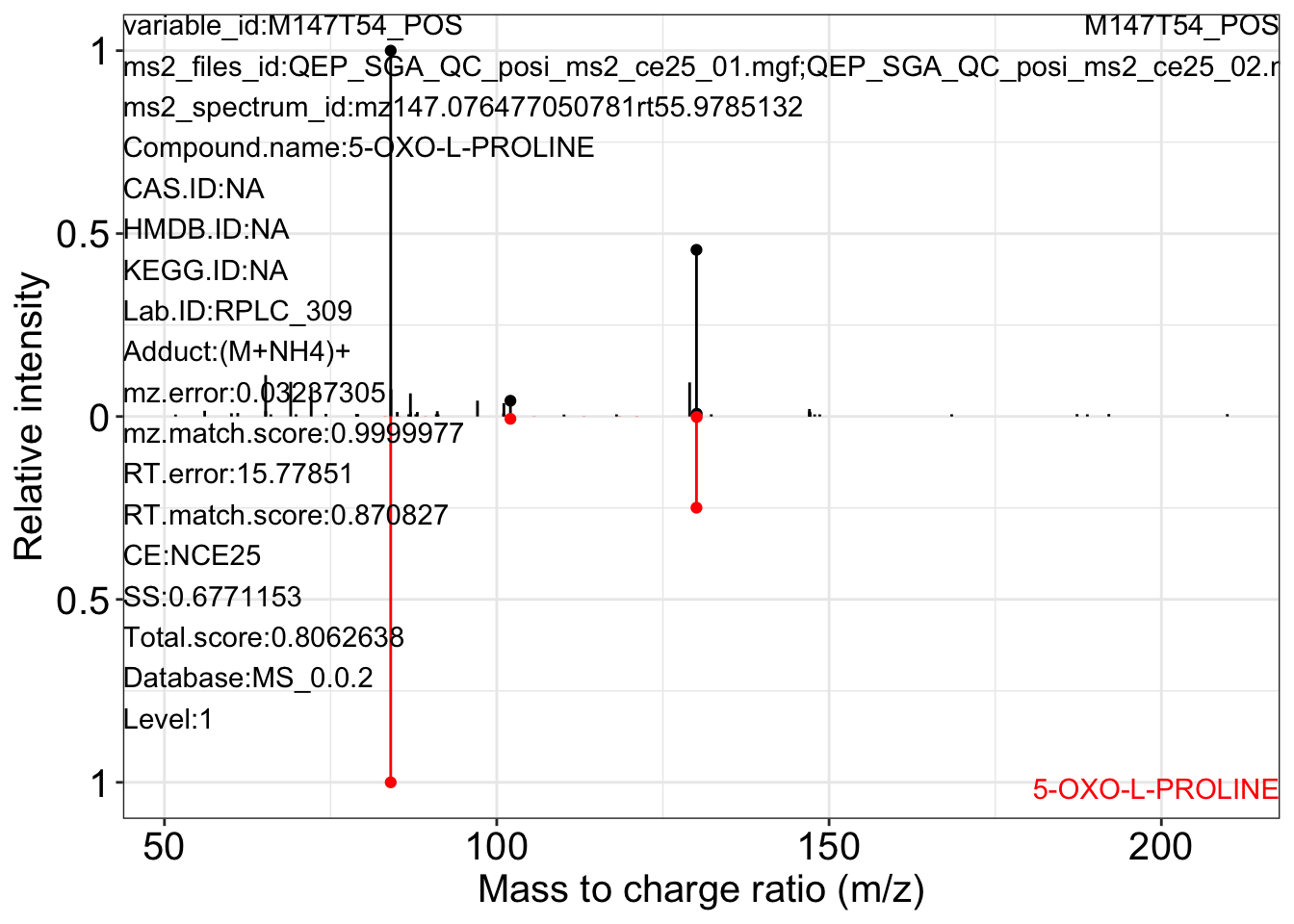
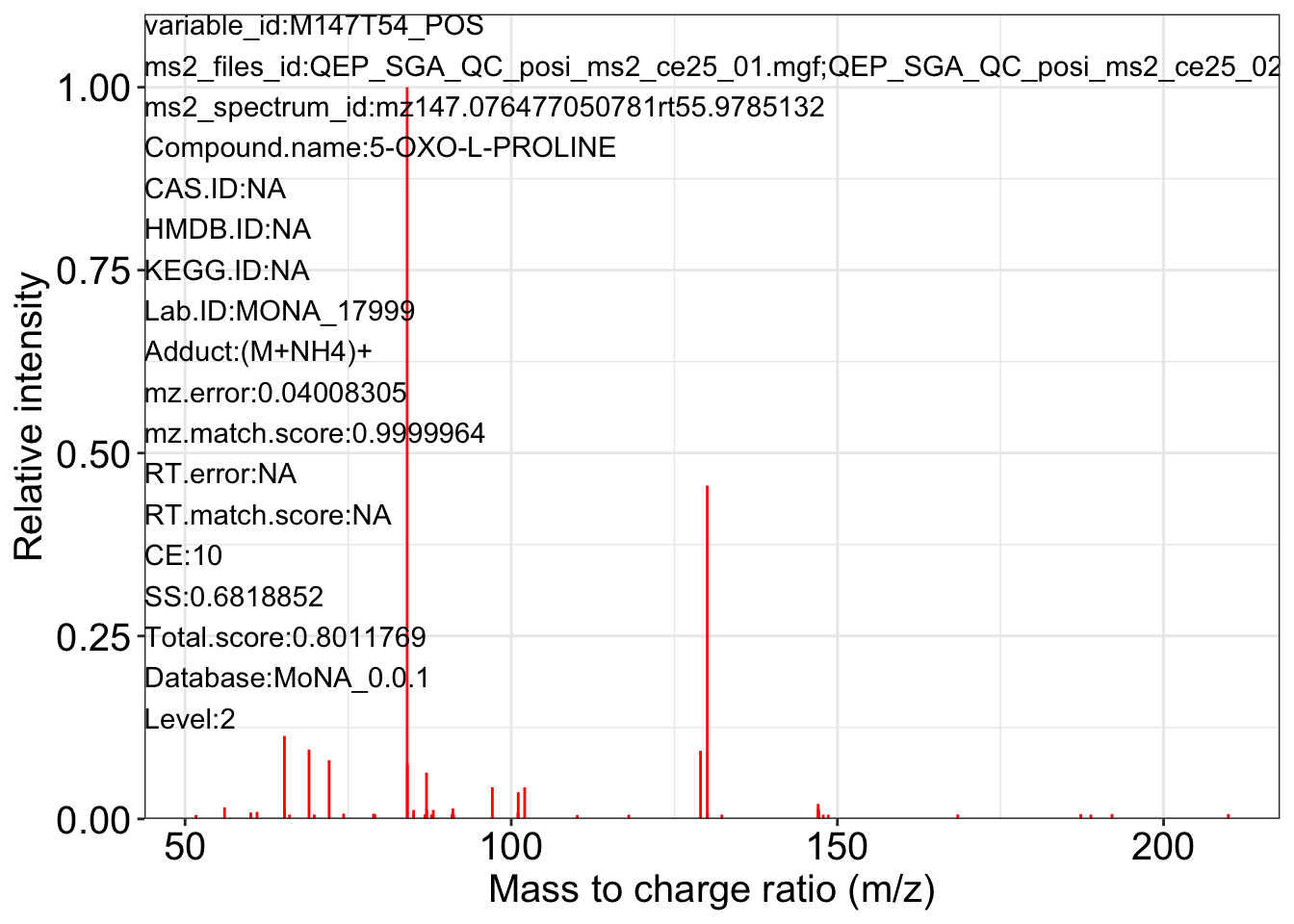
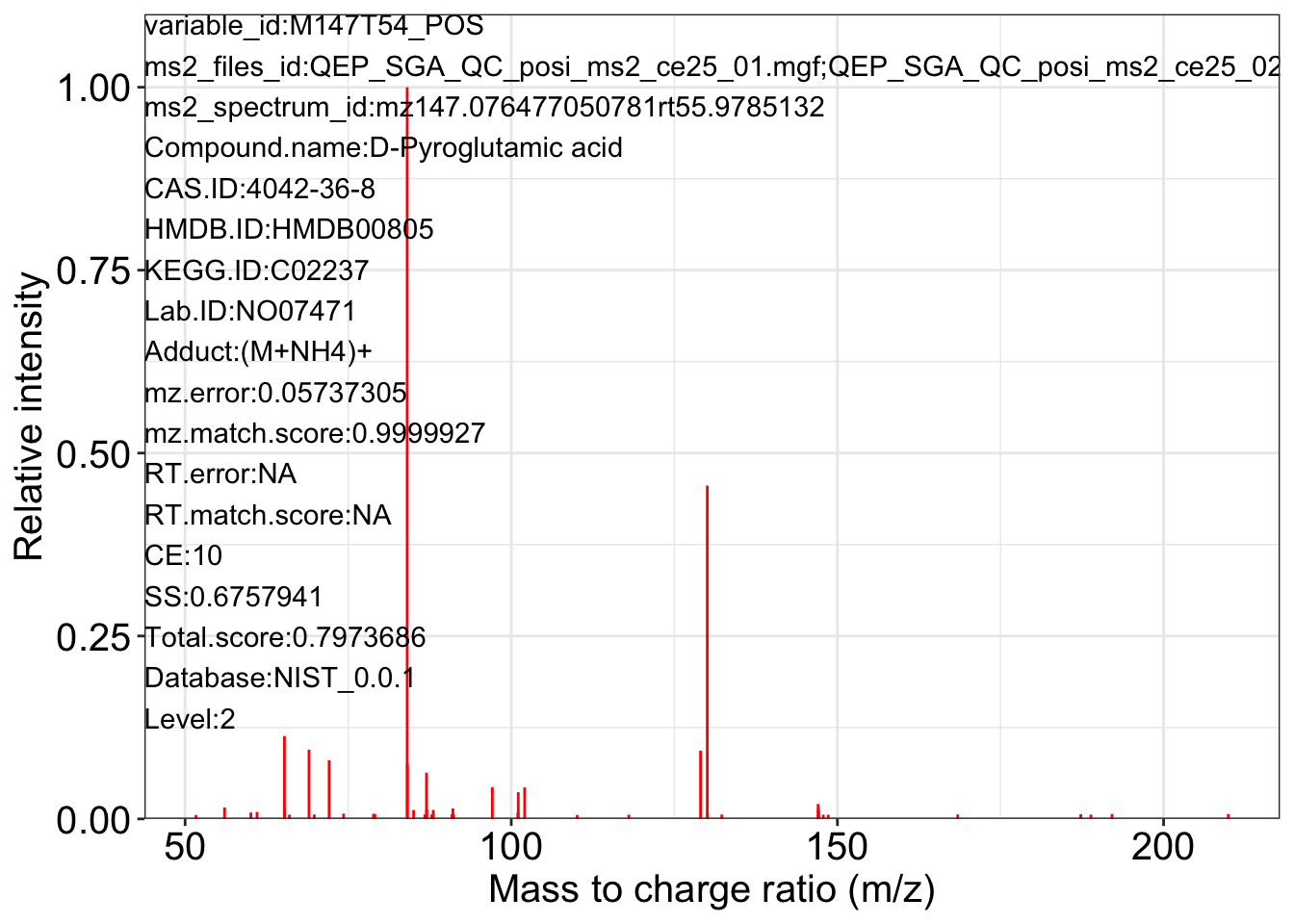
ms2_plot_mass_dataset(object = object_pos2,
variable_id = "M147T54_POS",
database = snyder_database_rplc0.0.3,
interactive_plot = FALSE)
#> database may be wrong.
#> database may be wrong.
#> $M147T54_POS_1
#>
#> $M147T54_POS_2
#>
#> $M147T54_POS_3
Save data for subsequent analysis.
save(object_pos2, file = "metabolite_annotation/object_pos2")
save(object_neg2, file = "metabolite_annotation/object_neg2")
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS Sonoma 14.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] metid_1.2.32 metpath_1.0.8 ComplexHeatmap_2.20.0
#> [4] mixOmics_6.28.0 lattice_0.22-6 MASS_7.3-61
#> [7] massstat_1.0.6 tidyr_1.3.1 ggfortify_0.4.17
#> [10] massqc_1.0.7 masscleaner_1.0.12 MSnbase_2.30.1
#> [13] ProtGenerics_1.36.0 S4Vectors_0.42.1 Biobase_2.64.0
#> [16] BiocGenerics_0.50.0 mzR_2.38.0 Rcpp_1.0.13
#> [19] xcms_4.2.3 BiocParallel_1.38.0 massprocesser_1.0.10
#> [22] ggplot2_3.5.1 dplyr_1.1.4 magrittr_2.0.3
#> [25] masstools_1.0.13 massdataset_1.0.34 tidymass_1.0.9
#>
#> loaded via a namespace (and not attached):
#> [1] fs_1.6.4 matrixStats_1.3.0
#> [3] bitops_1.0-8 fit.models_0.64
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 tools_4.4.1
#> [9] doRNG_1.8.6 backports_1.5.0
#> [11] utf8_1.2.4 R6_2.5.1
#> [13] lazyeval_0.2.2 GetoptLong_1.0.5
#> [15] withr_3.0.1 prettyunits_1.2.0
#> [17] gridExtra_2.3 preprocessCore_1.66.0
#> [19] cli_3.6.3 fastDummies_1.7.4
#> [21] labeling_0.4.3 sass_0.4.9
#> [23] mvtnorm_1.3-1 robustbase_0.99-4
#> [25] readr_2.1.5 randomForest_4.7-1.1
#> [27] proxy_0.4-27 pbapply_1.7-2
#> [29] foreign_0.8-87 rrcov_1.7-6
#> [31] MetaboCoreUtils_1.12.0 parallelly_1.38.0
#> [33] itertools_0.1-3 limma_3.60.4
#> [35] readxl_1.4.3 rstudioapi_0.16.0
#> [37] impute_1.78.0 generics_0.1.3
#> [39] shape_1.4.6.1 crosstalk_1.2.1
#> [41] zip_2.3.1 Matrix_1.7-0
#> [43] MALDIquant_1.22.3 fansi_1.0.6
#> [45] abind_1.4-5 lifecycle_1.0.4
#> [47] yaml_2.3.10 SummarizedExperiment_1.34.0
#> [49] SparseArray_1.4.8 crayon_1.5.3
#> [51] PSMatch_1.8.0 KEGGREST_1.44.1
#> [53] pillar_1.9.0 knitr_1.48
#> [55] GenomicRanges_1.56.1 rjson_0.2.22
#> [57] corpcor_1.6.10 codetools_0.2-20
#> [59] glue_1.7.0 pcaMethods_1.96.0
#> [61] data.table_1.16.0 remotes_2.5.0
#> [63] MultiAssayExperiment_1.30.3 vctrs_0.6.5
#> [65] png_0.1-8 cellranger_1.1.0
#> [67] gtable_0.3.5 cachem_1.1.0
#> [69] xfun_0.47 openxlsx_4.2.7
#> [71] S4Arrays_1.4.1 tidygraph_1.3.1
#> [73] pcaPP_2.0-5 ncdf4_1.23
#> [75] iterators_1.0.14 statmod_1.5.0
#> [77] robust_0.7-5 progress_1.2.3
#> [79] GenomeInfoDb_1.40.1 rprojroot_2.0.4
#> [81] bslib_0.8.0 affyio_1.74.0
#> [83] rpart_4.1.23 colorspace_2.1-1
#> [85] DBI_1.2.3 Hmisc_5.1-3
#> [87] nnet_7.3-19 tidyselect_1.2.1
#> [89] compiler_4.4.1 MassSpecWavelet_1.70.0
#> [91] htmlTable_2.4.3 DelayedArray_0.30.1
#> [93] plotly_4.10.4 bookdown_0.40
#> [95] checkmate_2.3.2 scales_1.3.0
#> [97] DEoptimR_1.1-3 affy_1.82.0
#> [99] stringr_1.5.1 digest_0.6.37
#> [101] rmarkdown_2.28 XVector_0.44.0
#> [103] htmltools_0.5.8.1 pkgconfig_2.0.3
#> [105] base64enc_0.1-3 MatrixGenerics_1.16.0
#> [107] highr_0.11 fastmap_1.2.0
#> [109] rlang_1.1.4 GlobalOptions_0.1.2
#> [111] htmlwidgets_1.6.4 UCSC.utils_1.0.0
#> [113] farver_2.1.2 jquerylib_0.1.4
#> [115] jsonlite_1.8.8 MsExperiment_1.6.0
#> [117] mzID_1.42.0 RCurl_1.98-1.16
#> [119] Formula_1.2-5 GenomeInfoDbData_1.2.12
#> [121] patchwork_1.2.0 munsell_0.5.1
#> [123] viridis_0.6.5 MsCoreUtils_1.16.1
#> [125] vsn_3.72.0 furrr_0.3.1
#> [127] stringi_1.8.4 ggraph_2.2.1
#> [129] zlibbioc_1.50.0 plyr_1.8.9
#> [131] parallel_4.4.1 listenv_0.9.1
#> [133] ggrepel_0.9.5 Biostrings_2.72.1
#> [135] MsFeatures_1.12.0 graphlayouts_1.1.1
#> [137] hms_1.1.3 Spectra_1.14.1
#> [139] circlize_0.4.16 igraph_2.0.3
#> [141] QFeatures_1.14.2 rngtools_1.5.2
#> [143] reshape2_1.4.4 XML_3.99-0.17
#> [145] evaluate_0.24.0 blogdown_1.19
#> [147] BiocManager_1.30.25 tzdb_0.4.0
#> [149] foreach_1.5.2 missForest_1.5
#> [151] tweenr_2.0.3 purrr_1.0.2
#> [153] polyclip_1.10-7 future_1.34.0
#> [155] clue_0.3-65 ggforce_0.4.2
#> [157] AnnotationFilter_1.28.0 e1071_1.7-14
#> [159] RSpectra_0.16-2 ggcorrplot_0.1.4.1
#> [161] viridisLite_0.4.2 class_7.3-22
#> [163] rARPACK_0.11-0 tibble_3.2.1
#> [165] memoise_2.0.1 ellipse_0.5.0
#> [167] IRanges_2.38.1 cluster_2.1.6
#> [169] globals_0.16.3 here_1.0.1