Data preparation
We just use the dataset which are from this step.
library(tidymass)
#> Registered S3 method overwritten by 'Hmisc':
#> method from
#> vcov.default fit.models
#> ── Attaching packages ────────────────────────────── tidymass 1.0.9 ──
#> ✔ massdataset 1.0.34 ✔ metid 1.2.33
#> ✔ massprocesser 1.0.10 ✔ masstools 1.0.13
#> ✔ masscleaner 1.0.12 ✔ dplyr 1.1.4
#> ✔ massqc 1.0.7 ✔ ggplot2 3.5.1
#> ✔ massstat 1.0.6 ✔ magrittr 2.0.3
#> ✔ metpath 1.0.8
load("data_cleaning/POS/object_pos")
load("data_cleaning/NEG/object_neg")
Change batch to character.
object_pos <-
object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
dplyr::mutate(batch = as.character(batch))
object_neg <-
object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
dplyr::mutate(batch = as.character(batch))
object_pos
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 10149 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 10149 x 3 data.frame]
#> 10149 variables:M70T73_POS M70T53_POS M70T183_POS ... M923T55_POS M992T641_POS
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 3 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 3 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:37:06
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:36:42
#> mutate ----------
#> Package Function.used Time
#> 1 massdataset mutate() 2024-09-25 19:53:23
object_neg
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 8804 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 8804 x 3 data.frame]
#> 8804 variables:M70T712_NEG M70T527_NEG M70T587_NEG ... M941T65_NEG M948T641_NEG
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 3 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 3 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:38:19
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:38:02
#> mutate ----------
#> Package Function.used Time
#> 1 massdataset mutate() 2024-09-25 19:53:23
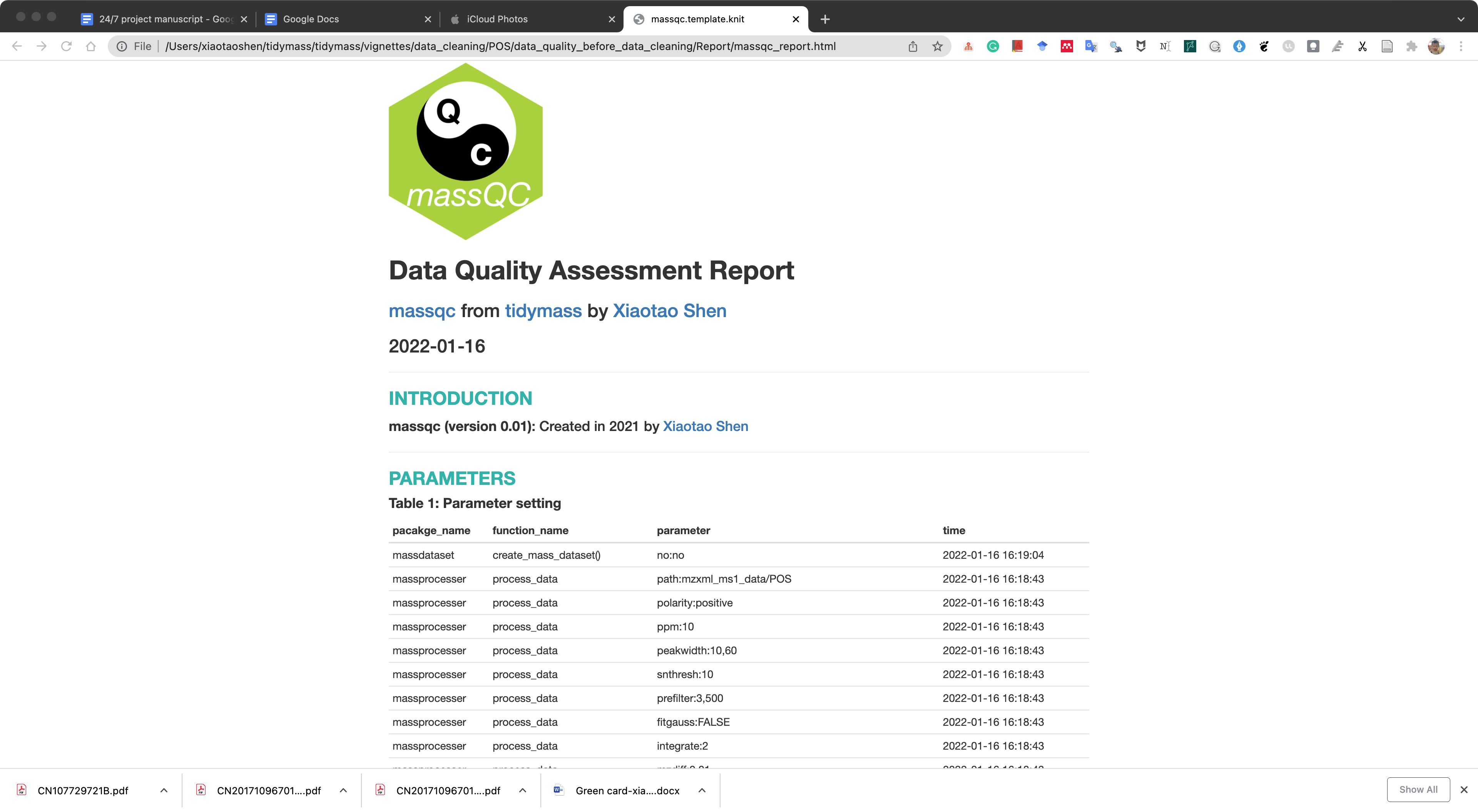
Data quality assessment before data cleaning
Here, we can use the massqc package to assess the data quality.
We can just use the massqc_report() function to get a html format quality assessment report.
massqc::massqc_report(object = object_pos,
path = "data_cleaning/POS/data_quality_before_data_cleaning")
A html format report and pdf figures will be placed in the folder data_cleaning/POS/data_quality_before_data_cleaning/Report.
The html report is below:
Negative mode.
massqc::massqc_report(object = object_neg,
path = "data_cleaning/NEG/data_quality_before_data_cleaning")
The PCA score plot is used to show the batch effect of positive and negative dataset.
Positive mode:
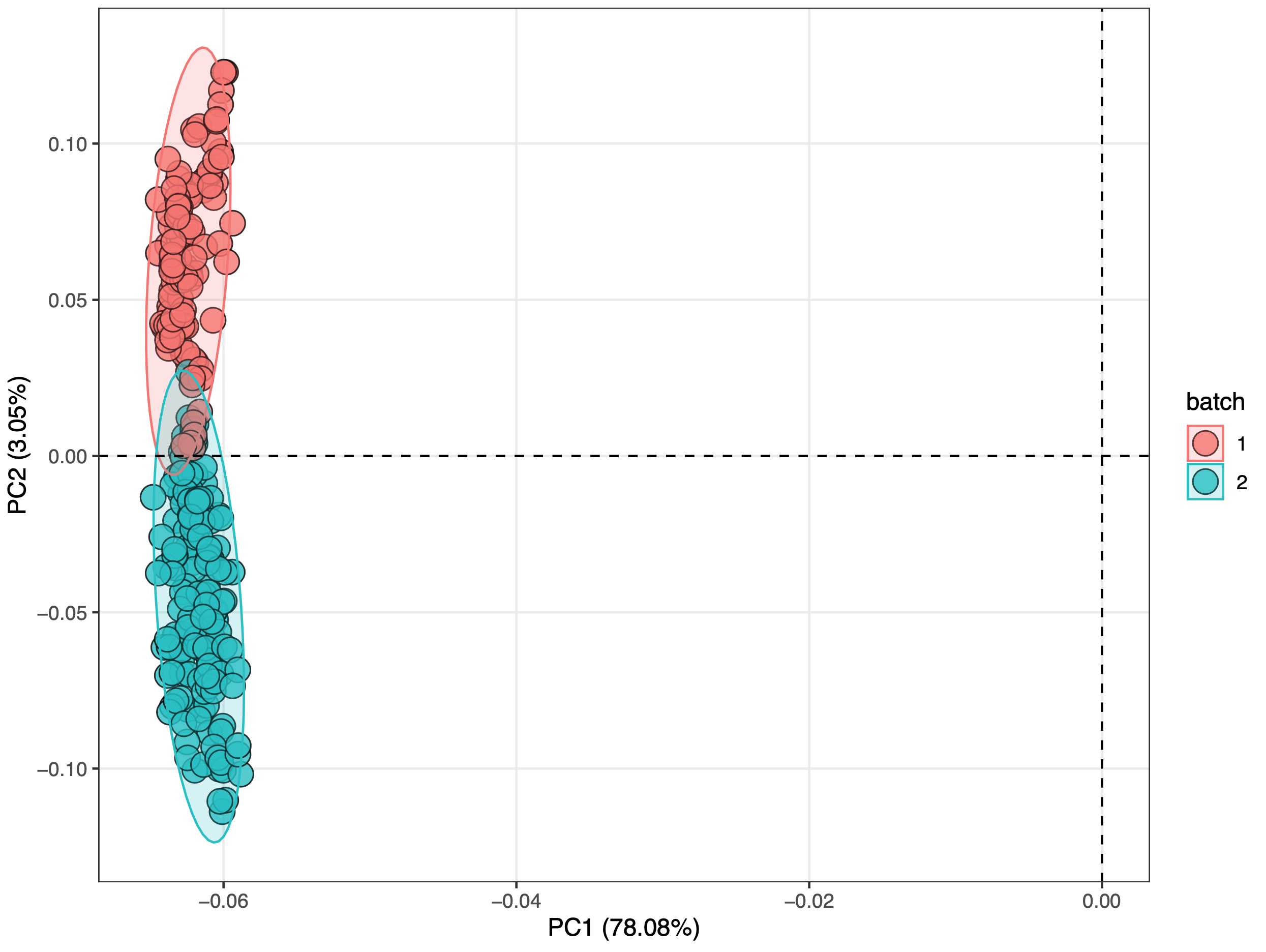
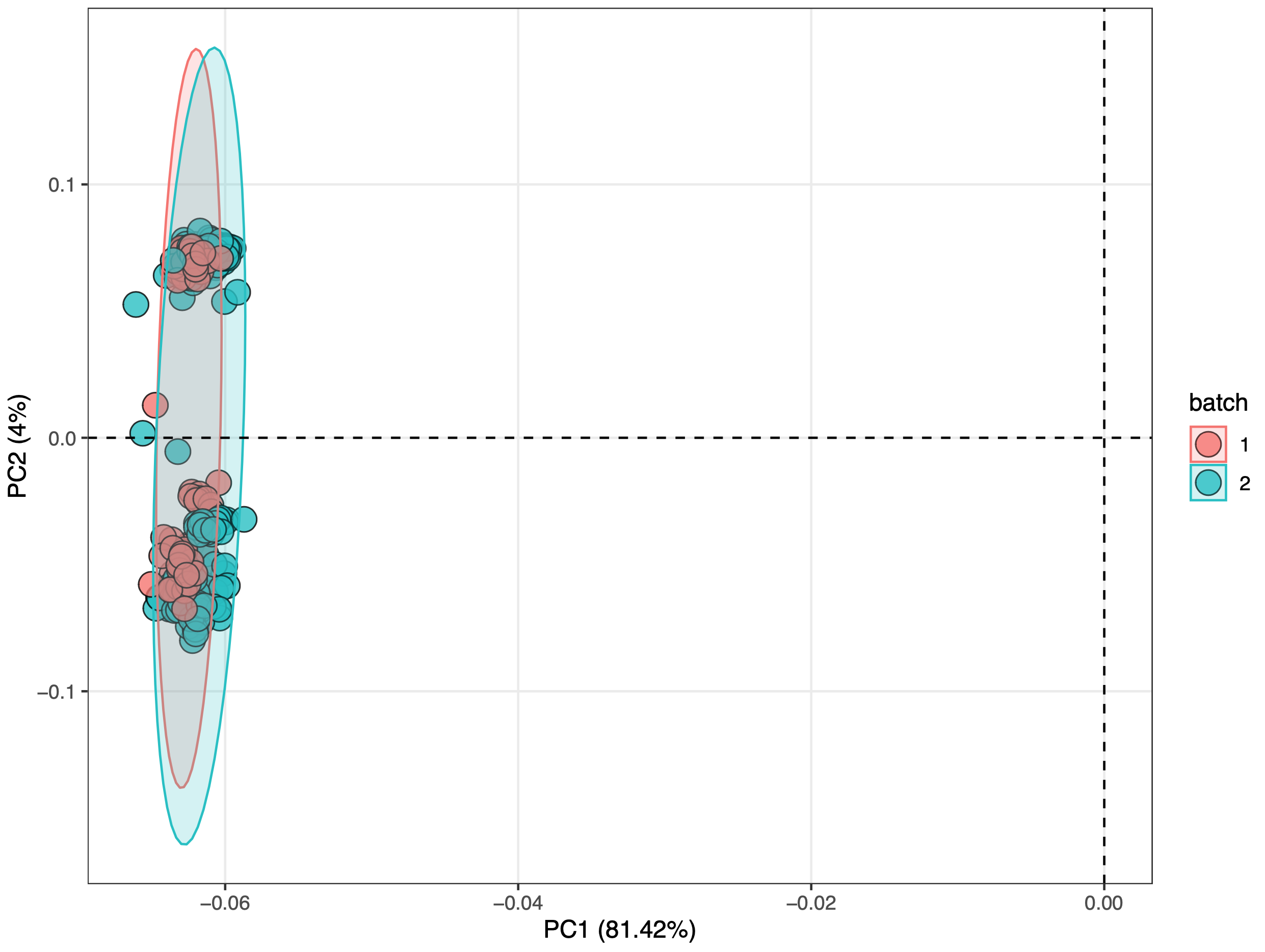
We can see that no matter in positive and negative mode, batch effect is serious.
Remove noisy metabolic features
Remove variables which have MVs in more than 20% QC samples and in at lest 50% samples in control group or case group.
Positive mode
object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
dplyr::count(group)
#> group n
#> 1 Case 110
#> 2 Control 110
#> 3 QC 39
MV percentage in QC samples.
show_variable_missing_values(object = object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
filter(class == "QC"),
percentage = TRUE) +
scale_size_continuous(range = c(0.01, 2))

qc_id =
object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
filter(class == "QC") %>%
pull(sample_id)
control_id =
object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
filter(group == "Control") %>%
pull(sample_id)
case_id =
object_pos %>%
activate_mass_dataset(what = "sample_info") %>%
filter(group == "Case") %>%
pull(sample_id)
object_pos =
object_pos %>%
mutate_variable_na_freq(according_to_samples = qc_id) %>%
mutate_variable_na_freq(according_to_samples = control_id) %>%
mutate_variable_na_freq(according_to_samples = case_id)
head(extract_variable_info(object_pos))
#> variable_id mz rt na_freq na_freq.1 na_freq.2
#> 1 M70T73_POS 70.04074 73.31705 0.28205128 0.6000000 0.4727273
#> 2 M70T53_POS 70.06596 52.78542 0.00000000 0.1454545 0.0000000
#> 3 M70T183_POS 70.19977 182.87981 0.00000000 0.6636364 0.7454545
#> 4 M70T527_POS 70.36113 526.76657 0.02564103 0.1818182 0.3000000
#> 5 M71T695_POS 70.56911 694.84592 0.02564103 0.6454545 0.5545455
#> 6 M71T183_POS 70.75034 182.77790 0.05128205 0.7272727 0.7909091
Remove variables.
object_pos <-
object_pos %>%
activate_mass_dataset(what = "variable_info") %>%
filter(na_freq < 0.2 & (na_freq.1 < 0.5 | na_freq.2 < 0.5))
object_pos
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 5101 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 5101 x 6 data.frame]
#> 5101 variables:M70T53_POS M70T527_POS M71T775_POS ... M836T610_POS M836T759_POS
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 6 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 5 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:37:06
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:36:42
#> mutate ----------
#> Package Function.used Time
#> 1 massdataset mutate() 2024-09-25 19:53:23
#> mutate_variable_na_freq ----------
#> Package Function.used Time
#> 1 massdataset mutate_variable_na_freq() 2024-09-25 19:53:25.443142
#> 2 massdataset mutate_variable_na_freq() 2024-09-25 19:53:25.465639
#> 3 massdataset mutate_variable_na_freq() 2024-09-25 19:53:25.505201
#> filter ----------
#> Package Function.used Time
#> 1 massdataset filter() 2024-09-25 19:53:25
Only 5,101 variables left.
Negative mode
object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
dplyr::count(group)
#> group n
#> 1 Case 110
#> 2 Control 110
#> 3 QC 39
MV percentage in QC samples.
show_variable_missing_values(object = object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
filter(class == "QC"),
percentage = TRUE) +
scale_size_continuous(range = c(0.01, 2))

qc_id =
object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
filter(class == "QC") %>%
pull(sample_id)
control_id =
object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
filter(group == "Control") %>%
pull(sample_id)
case_id =
object_neg %>%
activate_mass_dataset(what = "sample_info") %>%
filter(group == "Case") %>%
pull(sample_id)
object_neg =
object_neg %>%
mutate_variable_na_freq(according_to_samples = qc_id) %>%
mutate_variable_na_freq(according_to_samples = control_id) %>%
mutate_variable_na_freq(according_to_samples = case_id)
head(extract_variable_info(object_neg))
#> variable_id mz rt na_freq na_freq.1 na_freq.2
#> 1 M70T712_NEG 70.05911 711.97894 0.05128205 0.109090909 0.018181818
#> 2 M70T527_NEG 70.13902 526.85416 0.33333333 0.509090909 0.618181818
#> 3 M70T587_NEG 70.31217 587.25330 0.00000000 0.009090909 0.009090909
#> 4 M70T47_NEG 70.33835 46.57537 0.84615385 0.936363636 0.090909091
#> 5 M71T587_NEG 70.56315 587.02570 0.17948718 0.145454545 0.163636364
#> 6 M71T641_NEG 70.70657 641.16672 0.10256410 0.063636364 0.072727273
Remove variables.
object_neg <-
object_neg %>%
activate_mass_dataset(what = "variable_info") %>%
filter(na_freq < 0.2 & (na_freq.1 < 0.5 | na_freq.2 < 0.5))
object_neg
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 4104 x 259 data.frame]
#> 2.sample_info:[ 259 x 6 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 4104 x 6 data.frame]
#> 4104 variables:M70T712_NEG M70T587_NEG M71T587_NEG ... M884T57_NEG M899T56_NEG
#> 4.sample_info_note:[ 6 x 2 data.frame]
#> 5.variable_info_note:[ 6 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 5 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:38:19
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:38:02
#> mutate ----------
#> Package Function.used Time
#> 1 massdataset mutate() 2024-09-25 19:53:23
#> mutate_variable_na_freq ----------
#> Package Function.used Time
#> 1 massdataset mutate_variable_na_freq() 2024-09-25 19:53:26.396638
#> 2 massdataset mutate_variable_na_freq() 2024-09-25 19:53:26.426459
#> 3 massdataset mutate_variable_na_freq() 2024-09-25 19:53:26.447222
#> filter ----------
#> Package Function.used Time
#> 1 massdataset filter() 2024-09-25 19:53:26
4104 features left.
Filter outlier samples
We can use the detect_outlier() to find the outlier samples.
More information about how to detect outlier samples can be found here.
Positive mode
massdataset::show_sample_missing_values(object = object_pos,
color_by = "group",
order_by = "injection.order",
percentage = TRUE) +
theme(axis.text.x = element_text(size = 2)) +
scale_size_continuous(range = c(0.1, 2)) +
ggsci::scale_color_aaas()

Detect outlier samples.
outlier_samples =
object_pos %>%
`+`(1) %>%
log(2) %>%
scale() %>%
detect_outlier()
outlier_samples
#> --------------------
#> masscleaner
#> --------------------
#> 1 according_to_na : 0 outlier samples.
#> 2 according_to_pc_sd : 0 outlier samples.
#> 3 according_to_pc_mad : 0 outlier samples.
#> 4 accordint_to_distance : 0 outlier samples.
#> extract all outlier table using extract_outlier_table()
outlier_table <-
extract_outlier_table(outlier_samples)
outlier_table %>%
head()
#> according_to_na pc_sd pc_mad accordint_to_distance
#> sample_06 FALSE FALSE FALSE FALSE
#> sample_103 FALSE FALSE FALSE FALSE
#> sample_11 FALSE FALSE FALSE FALSE
#> sample_112 FALSE FALSE FALSE FALSE
#> sample_117 FALSE FALSE FALSE FALSE
#> sample_12 FALSE FALSE FALSE FALSE
outlier_table %>%
apply(1, function(x){
sum(x)
}) %>%
`>`(0) %>%
which()
#> named integer(0)
No outlier samples in positive mode.
Negative mode
massdataset::show_sample_missing_values(object = object_neg,
color_by = "group",
order_by = "injection.order",
percentage = TRUE) +
theme(axis.text.x = element_text(size = 2)) +
scale_size_continuous(range = c(0.1, 2)) +
ggsci::scale_color_aaas()

Detect outlier samples.
outlier_samples =
object_neg %>%
`+`(1) %>%
log(2) %>%
scale() %>%
detect_outlier()
outlier_samples
#> --------------------
#> masscleaner
#> --------------------
#> 1 according_to_na : 0 outlier samples.
#> 2 according_to_pc_sd : 0 outlier samples.
#> 3 according_to_pc_mad : 0 outlier samples.
#> 4 accordint_to_distance : 0 outlier samples.
#> extract all outlier table using extract_outlier_table()
outlier_table <-
extract_outlier_table(outlier_samples)
outlier_table %>%
head()
#> according_to_na pc_sd pc_mad accordint_to_distance
#> sample_06 FALSE FALSE FALSE FALSE
#> sample_103 FALSE FALSE FALSE FALSE
#> sample_11 FALSE FALSE FALSE FALSE
#> sample_112 FALSE FALSE FALSE FALSE
#> sample_117 FALSE FALSE FALSE FALSE
#> sample_12 FALSE FALSE FALSE FALSE
outlier_table %>%
apply(1, function(x){
sum(x)
}) %>%
`>`(0) %>%
which()
#> named integer(0)
No outlier samples in negative mode.
Missing value imputation
get_mv_number(object_pos)
#> [1] 148965
object_pos <-
impute_mv(object = object_pos, method = "knn")
#> Cluster size 4983 broken into 88 4895
#> Done cluster 88
#> Cluster size 4895 broken into 4497 398
#> Cluster size 4497 broken into 3737 760
#> Cluster size 3737 broken into 2703 1034
#> Cluster size 2703 broken into 1706 997
#> Cluster size 1706 broken into 1240 466
#> Done cluster 1240
#> Done cluster 466
#> Done cluster 1706
#> Done cluster 997
#> Done cluster 2703
#> Done cluster 1034
#> Done cluster 3737
#> Done cluster 760
#> Done cluster 4497
#> Done cluster 398
#> Done cluster 4895
get_mv_number(object_pos)
#> [1] 0
get_mv_number(object_neg)
#> [1] 146427
object_neg <-
impute_mv(object = object_neg, method = "knn")
#> Cluster size 4006 broken into 3965 41
#> Cluster size 3965 broken into 3743 222
#> Cluster size 3743 broken into 505 3238
#> Done cluster 505
#> Cluster size 3238 broken into 2519 719
#> Cluster size 2519 broken into 1721 798
#> Cluster size 1721 broken into 676 1045
#> Done cluster 676
#> Done cluster 1045
#> Done cluster 1721
#> Done cluster 798
#> Done cluster 2519
#> Done cluster 719
#> Done cluster 3238
#> Done cluster 3743
#> Done cluster 222
#> Done cluster 3965
#> Done cluster 41
get_mv_number(object_neg)
#> [1] 0
Data normalization and integration
Positive mode
object_pos <-
normalize_data(object_pos, method = "median")
object_pos2 <-
integrate_data(object_pos, method = "subject_median")
object_pos2 %>%
`+`(1) %>%
log(2) %>%
massqc::massqc_pca(color_by = "batch", line = FALSE)

Negative mode
object_neg <-
normalize_data(object_neg, method = "median")
object_neg2 <-
integrate_data(object_neg, method = "subject_median")
object_neg2 %>%
`+`(1) %>%
log(2) %>%
massqc::massqc_pca(color_by = "batch", line = FALSE)

Save data for subsequent analysis.
save(object_pos2, file = "data_cleaning/POS/object_pos2")
save(object_neg2, file = "data_cleaning/NEG/object_neg2")
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.0
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] metid_1.2.33 metpath_1.0.8 ComplexHeatmap_2.20.0
#> [4] mixOmics_6.28.0 lattice_0.22-6 MASS_7.3-61
#> [7] massstat_1.0.6 tidyr_1.3.1 ggfortify_0.4.17
#> [10] massqc_1.0.7 masscleaner_1.0.12 MSnbase_2.30.1
#> [13] ProtGenerics_1.36.0 S4Vectors_0.42.1 Biobase_2.64.0
#> [16] BiocGenerics_0.50.0 mzR_2.38.0 Rcpp_1.0.13
#> [19] xcms_4.2.3 BiocParallel_1.38.0 massprocesser_1.0.10
#> [22] ggplot2_3.5.1 dplyr_1.1.4 magrittr_2.0.3
#> [25] masstools_1.0.13 massdataset_1.0.34 tidymass_1.0.9
#>
#> loaded via a namespace (and not attached):
#> [1] fs_1.6.4 matrixStats_1.3.0
#> [3] bitops_1.0-8 fit.models_0.64
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 ggsci_3.2.0
#> [9] tools_4.4.1 doRNG_1.8.6
#> [11] backports_1.5.0 utf8_1.2.4
#> [13] R6_2.5.1 lazyeval_0.2.2
#> [15] GetoptLong_1.0.5 withr_3.0.1
#> [17] prettyunits_1.2.0 gridExtra_2.3
#> [19] preprocessCore_1.66.0 cli_3.6.3
#> [21] fastDummies_1.7.4 labeling_0.4.3
#> [23] sass_0.4.9 mvtnorm_1.3-1
#> [25] robustbase_0.99-4 readr_2.1.5
#> [27] randomForest_4.7-1.1 proxy_0.4-27
#> [29] pbapply_1.7-2 systemfonts_1.1.0
#> [31] foreign_0.8-87 svglite_2.1.3
#> [33] rrcov_1.7-6 MetaboCoreUtils_1.12.0
#> [35] parallelly_1.38.0 itertools_0.1-3
#> [37] limma_3.60.4 readxl_1.4.3
#> [39] rstudioapi_0.16.0 impute_1.78.0
#> [41] generics_0.1.3 shape_1.4.6.1
#> [43] zip_2.3.1 Matrix_1.7-0
#> [45] MALDIquant_1.22.3 fansi_1.0.6
#> [47] abind_1.4-5 lifecycle_1.0.4
#> [49] yaml_2.3.10 SummarizedExperiment_1.34.0
#> [51] SparseArray_1.4.8 crayon_1.5.3
#> [53] PSMatch_1.8.0 KEGGREST_1.44.1
#> [55] pillar_1.9.0 knitr_1.48
#> [57] GenomicRanges_1.56.1 rjson_0.2.22
#> [59] corpcor_1.6.10 codetools_0.2-20
#> [61] glue_1.7.0 pcaMethods_1.96.0
#> [63] data.table_1.16.0 remotes_2.5.0
#> [65] MultiAssayExperiment_1.30.3 vctrs_0.6.5
#> [67] png_0.1-8 cellranger_1.1.0
#> [69] gtable_0.3.5 cachem_1.1.0
#> [71] xfun_0.47 openxlsx_4.2.7
#> [73] S4Arrays_1.4.1 tidygraph_1.3.1
#> [75] pcaPP_2.0-5 ncdf4_1.23
#> [77] iterators_1.0.14 statmod_1.5.0
#> [79] robust_0.7-5 progress_1.2.3
#> [81] GenomeInfoDb_1.40.1 rprojroot_2.0.4
#> [83] bslib_0.8.0 affyio_1.74.0
#> [85] rpart_4.1.23 colorspace_2.1-1
#> [87] DBI_1.2.3 Hmisc_5.1-3
#> [89] nnet_7.3-19 tidyselect_1.2.1
#> [91] compiler_4.4.1 MassSpecWavelet_1.70.0
#> [93] htmlTable_2.4.3 DelayedArray_0.30.1
#> [95] plotly_4.10.4 bookdown_0.40
#> [97] checkmate_2.3.2 scales_1.3.0
#> [99] DEoptimR_1.1-3 affy_1.82.0
#> [101] stringr_1.5.1 digest_0.6.37
#> [103] rmarkdown_2.28 XVector_0.44.0
#> [105] htmltools_0.5.8.1 pkgconfig_2.0.3
#> [107] base64enc_0.1-3 MatrixGenerics_1.16.0
#> [109] highr_0.11 fastmap_1.2.0
#> [111] rlang_1.1.4 GlobalOptions_0.1.2
#> [113] htmlwidgets_1.6.4 UCSC.utils_1.0.0
#> [115] farver_2.1.2 jquerylib_0.1.4
#> [117] jsonlite_1.8.8 MsExperiment_1.6.0
#> [119] mzID_1.42.0 RCurl_1.98-1.16
#> [121] Formula_1.2-5 GenomeInfoDbData_1.2.12
#> [123] patchwork_1.2.0 munsell_0.5.1
#> [125] viridis_0.6.5 MsCoreUtils_1.16.1
#> [127] vsn_3.72.0 furrr_0.3.1
#> [129] stringi_1.8.4 ggraph_2.2.1
#> [131] zlibbioc_1.50.0 plyr_1.8.9
#> [133] parallel_4.4.1 listenv_0.9.1
#> [135] ggrepel_0.9.5 Biostrings_2.72.1
#> [137] MsFeatures_1.12.0 graphlayouts_1.1.1
#> [139] hms_1.1.3 Spectra_1.14.1
#> [141] circlize_0.4.16 igraph_2.0.3
#> [143] QFeatures_1.14.2 rngtools_1.5.2
#> [145] reshape2_1.4.4 XML_3.99-0.17
#> [147] evaluate_0.24.0 blogdown_1.19
#> [149] BiocManager_1.30.25 tzdb_0.4.0
#> [151] foreach_1.5.2 missForest_1.5
#> [153] tweenr_2.0.3 purrr_1.0.2
#> [155] polyclip_1.10-7 future_1.34.0
#> [157] clue_0.3-65 ggforce_0.4.2
#> [159] AnnotationFilter_1.28.0 e1071_1.7-14
#> [161] RSpectra_0.16-2 ggcorrplot_0.1.4.1
#> [163] viridisLite_0.4.2 class_7.3-22
#> [165] rARPACK_0.11-0 tibble_3.2.1
#> [167] memoise_2.0.1 ellipse_0.5.0
#> [169] IRanges_2.38.1 cluster_2.1.6
#> [171] globals_0.16.3 here_1.0.1