Introduction
If you want to construct databases for metid, you can use the massDatabase package. massdatabase is an R package that operates the online public databases and combines with other tools for streamlined compound annotation and pathway enrichment analysis. massdatabase is a flexible, simple, and powerful tool that can be installed on all platforms, allowing the users to leverage all the online public databases for biological function mining.
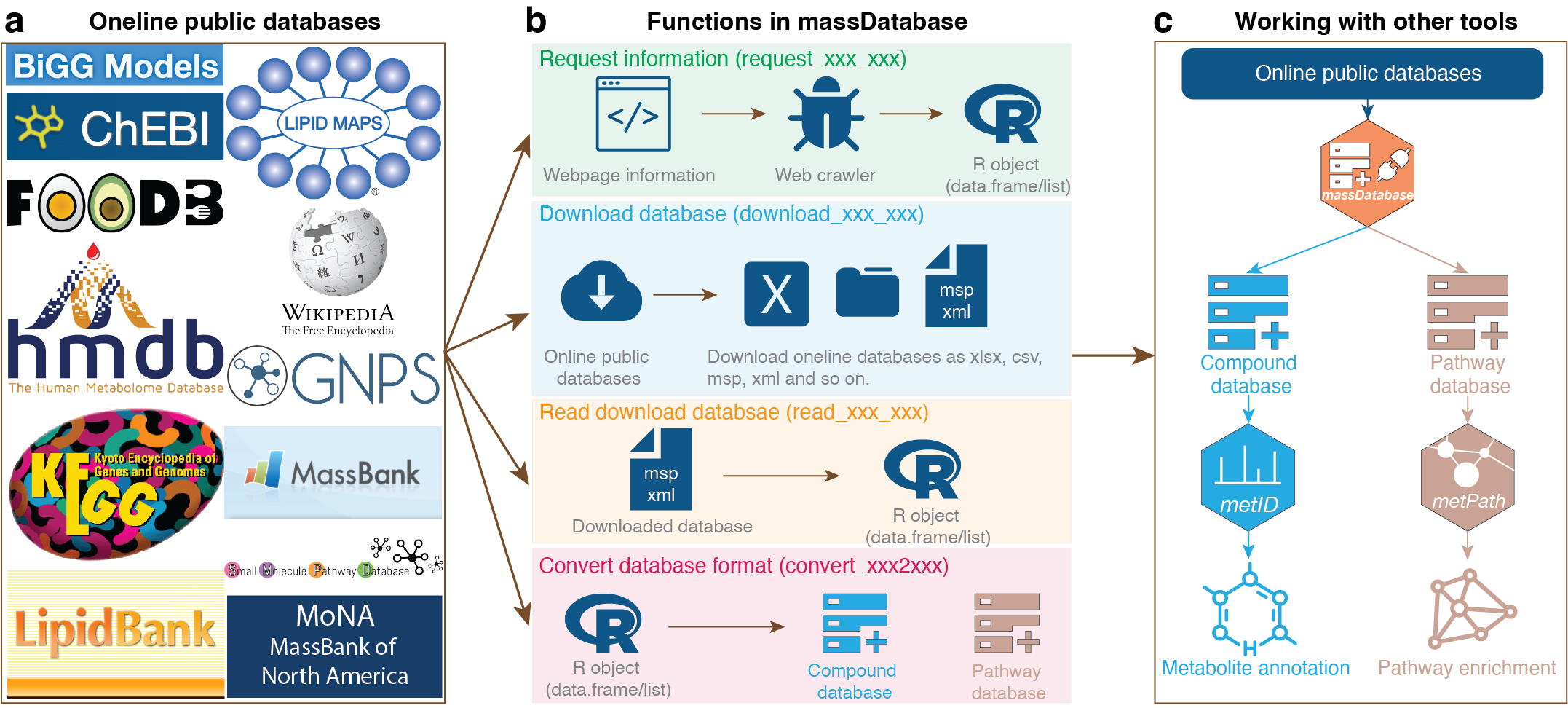
Install massdatabase
You can install massdatabase from GitLab.
if(!require(remotes)){
install.packages("remotes")
}
remotes::install_gitlab("tidymass/massdatabase")
or GitHub
remotes::install_github("tidymass/massdatabase")
or tidymass.org
source("https://www.tidymass.org/tidymass-packages/install_tidymass.txt")
install_tidymass(from = "tidymass.org", which_package = "massdatabase")
BIGG database
BIGG modelis a knowledge base of genome-scale metabolic network reconstructions.
Download the BIGG universal metabolite database:
library(massdatabase)
#> massdatabase 1.0.10 (2024-09-25 19:56:56.178093)
#>
#> Attaching package: 'massdatabase'
#> The following object is masked _by_ 'package:masstools':
#>
#> show_progresser
download_bigg_universal_metabolite(path = "database_construction/",
sleep = 1)
Then read and convert it to databaseClass format.
data <-
read_bigg_universal_metabolite(path = ".")
It may take a while to download the database.
Then convert it to databaseClass format.
bigg_database <-
convert_bigg_universal2metid(data = data, path = ".")
bigg_database
You can save the database to use later.
save(bigg_database, file = "database_construction/bigg_database")
ChEBI database
Chemical Entities of Biological Interest (ChEBI) is a freely available dictionary of molecular entities focused on ‘small’ chemical compounds.
Download the ChEBI compound database:
library(massdatabase)
download_chebi_compound(path = "database_construction/")
Then read and convert it to databaseClass format.
data <-
read_chebi_compound(path = "database_construction")
#> Reading data...
Then convert it to databaseClass format.
chebi_database <-
convert_chebi2metid(data = data, path = "database_construction")
#> No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmp1C3pOK/file16ed65065568b
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmp1C3pOK/file16ed65065568b
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
chebi_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:CHEBI
#> Link:https://www.ebi.ac.uk/chebi/init.do
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 34 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 152379 metabolites in total.
#> 0 metabolites with spectra in positive mode.
#> 0 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:0
#>
#> Collision energy in negative mode:
#> Total number:0
#>
You can save the database to use later.
save(chebi_database, file = "database_construction/chebi_database")
FooDB database
FooDB is the world’s largest and most comprehensive resource on food constituents, chemistry and biology.
Download the FooDB database:
library(massdatabase)
download_foodb_compound(compound_id = "all", path = "database_construction/")
It may take a while to download the database.
Then read and convert it to databaseClass format.
data <-
read_foodb_compound(path = "database_construction")
Then convert it to databaseClass format.
foodb_dataabse <-
convert_foodb2metid(data = data, path = "database_construction")
foodb_dataabse
You can save the database to use later.
save(foodb_dataabse, file = "database_construction/foodb_dataabse")
GNPS database
The GNPS GNPS is a web-based mass spectrometry ecosystem that aims to be an open-access knowledge base for community-wide organization and sharing of raw, processed, or annotated fragmentation mass spectrometry data (MS/MS). GNPS aids in identification and discovery throughout the entire life cycle of data; from initial data acquisition/analysis to post publication.
Download the database:
library(massdatabase)
download_gnps_spectral_library(gnps_library = "HMDB",
path = "database_construction")
The argument gnps_library should be one of the GNPS Library one this website.
https://gnps-external.ucsd.edu/gnpslibrary
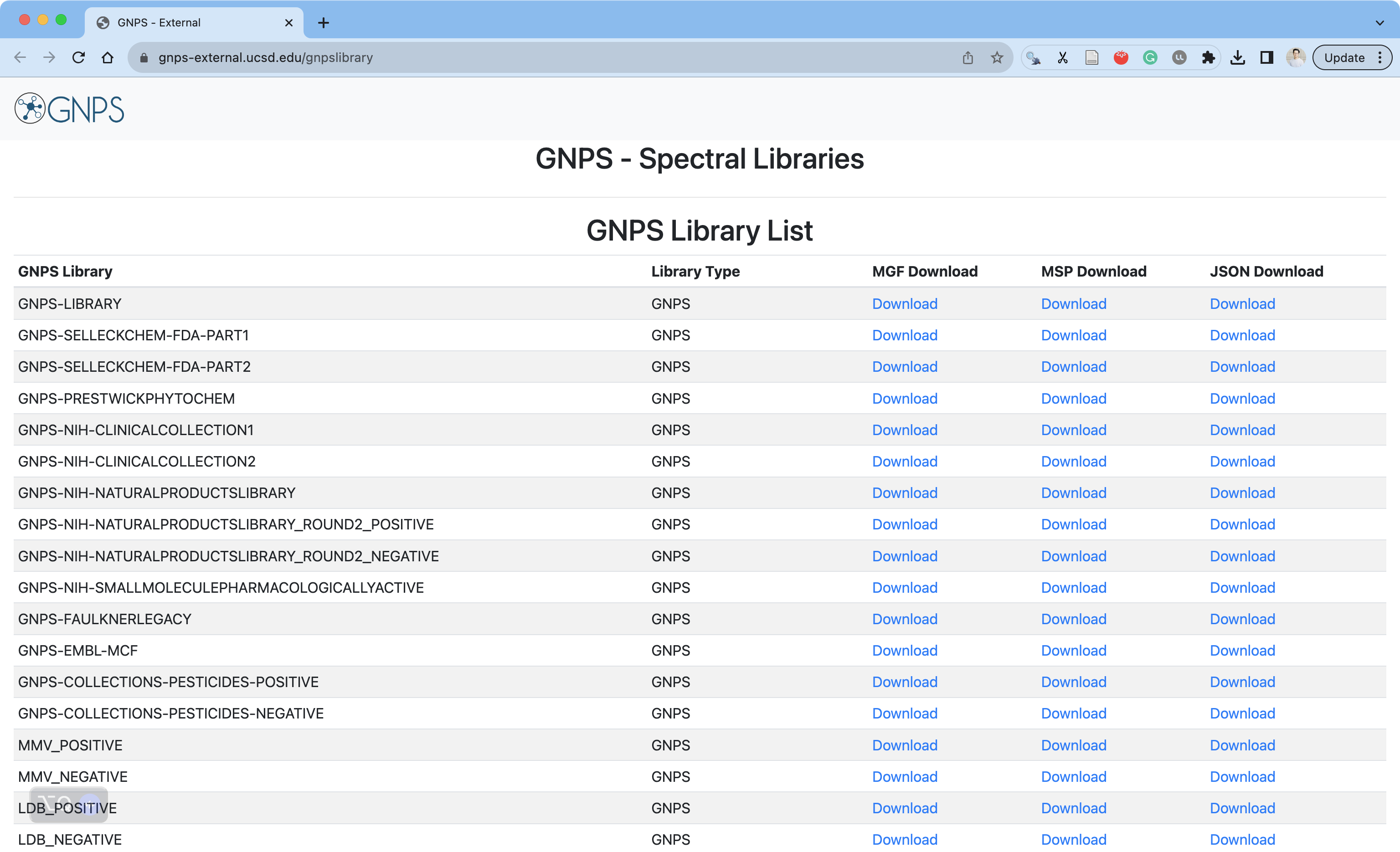
It may take a while to download the database.
Then read and convert it to databaseClass format.
data <-
read_msp_data_gnps(file = "database_construction/HMDB.msp")
#>
indexing HMDB.msp [=======================================] 339.58GB/s, eta: 0s
#> 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
Then convert it to databaseClass format.
gnps_database <-
convert_gnps2metid(data = data, path = "database_construction/")
#> Extracting MS1 inforamtion...
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Done.
#> Extracting MS2 inforamtion...
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Done.
#> Organizing...
#> Calculating accurate mass...
#> Done.
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmp1C3pOK/file16ed658ce7af6
#>
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmp1C3pOK/file16ed658ce7af6
#>
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
gnps_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:GNPS
#> Link:https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 25 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 2235 metabolites in total.
#> 1907 metabolites with spectra in positive mode.
#> 328 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:1
#> Unknown_1
#> Collision energy in negative mode:
#> Total number:1
#> Unknown_1
You can save the database to use later.
save(gnps_database, file = "database_construction/gnps_database")
KEGG database
KEGG is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies.
Download the database:
library(massdatabase)
download_kegg_compound(path = "database_construction/")
It may take a while to download the database.
Then read and convert it to databaseClass format.
data <-
read_kegg_compound(path = "database_construction/")
Then convert it to databaseClass format.
kegg_database <-
convert_kegg2metid(data = data, path = "database_construction")
kegg_database
You can save the database to use later.
save(kegg_database, file = "database_construction/kegg_database")
LipidMaps
LIPID MAPS Lipidomics Gateway was created in 2003 via an NIH “Glue Grant” to provide access to lipid nomenclature, databases, tools, protocols, standards, tutorials, meetings, publications, and other resources and serving the international lipid research community.
Download the database:
library(massdatabase)
download_lipidmaps_lipid(path = "database_construction")
It may take a while to download the database.
Uncompress the downloaded zip file.
unzip(zipfile = "database_construction/LMSD.sdf.zip",
exdir = "database_construction/")
Then read and convert it to databaseClass format.
data <-
read_sdf_data_lipidmaps(file = "database_construction/structures.sdf")
#> Reading data, it may take a while...
#> Done.
#> Organizing...
#> Done.
Then convert it to databaseClass format.
lipidmaps_database <-
convert_lipidmaps2metid(data = data, path = "database_construction/")
#> Organizing...
#> No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//RtmpeW0OGA/file171164ab3a0b1
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//RtmpeW0OGA/file171164ab3a0b1
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
lipidmaps_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:LIPIDMAPS
#> Link:https://www.lipidmaps.org/
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 27 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 47500 metabolites in total.
#> 0 metabolites with spectra in positive mode.
#> 0 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:0
#>
#> Collision energy in negative mode:
#> Total number:0
#>
You can save the database to use later.
save(lipidmaps_database, file = "database_construction/lipidmaps_database")
MassBank
MassBank is a community effort and you are invited to contribute. Please refer to our contributor documentation and get in touch via github or email.
Download the database:
library(massdatabase)
download_massbank_compound(source = "nist", path = "database_construction/")
It may take a while to download the database.
Then read and convert it to databaseClass format.
data <- read_msp_data_massbank(file = "database_construction/massbank_compound/MassBank_NIST.msp")
#> 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
It may take a while to read the data.
Then convert it to databaseClass format.
massbank_database <-
convert_massbank2metid_nist(data = data, path = "database_construction/")
It may take a while to convert the data.
massbank_database
You can save the database to use later.
save(massbank_database, file = "database_construction/massbank_database")
SMPDB
SMPDB is an interactive, visual database containing more than 30000 small molecule pathways found in humans only.
Download the database:
library(massdatabase)
download_smpdb_pathway(path = "database_construction/")
It may take a while to download the database.
Then read and convert it to databaseClass format.
data <-
read_smpdb_pathway(path = "database_construction/", only_primarity_pathway = TRUE)
Then convert it to databaseClass format.
smpdb_pathway_database <-
convert_smpdb2metpath(data = data, path = "database_construction/")
smpdb_pathway_database
You can save the database to use later.
save(smpdb_pathway_database, file = "database_construction/smpdb_pathway_database")
Citation
If you use massdatabase in your studies, please cite this paper:
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.0
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] MSnbase_2.30.1 ProtGenerics_1.36.0 S4Vectors_0.42.1
#> [4] mzR_2.38.0 Rcpp_1.0.13 Biobase_2.64.0
#> [7] BiocGenerics_0.50.0 massdataset_1.0.34 metid_1.2.33
#> [10] metpath_1.0.8 ggplot2_3.5.1 dplyr_1.1.4
#> [13] magrittr_2.0.3 masstools_1.0.13 massdatabase_1.0.10
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-8 tibble_3.2.1
#> [3] cellranger_1.1.0 polyclip_1.10-7
#> [5] preprocessCore_1.66.0 XML_3.99-0.17
#> [7] lifecycle_1.0.4 doParallel_1.0.17
#> [9] rprojroot_2.0.4 vroom_1.6.5
#> [11] globals_0.16.3 lattice_0.22-6
#> [13] MASS_7.3-61 MultiAssayExperiment_1.30.3
#> [15] openxlsx_4.2.7 limma_3.60.4
#> [17] plotly_4.10.4 sass_0.4.9
#> [19] rmarkdown_2.28 jquerylib_0.1.4
#> [21] yaml_2.3.10 remotes_2.5.0
#> [23] zip_2.3.1 MsCoreUtils_1.16.1
#> [25] pbapply_1.7-2 DBI_1.2.3
#> [27] RColorBrewer_1.1-3 abind_1.4-5
#> [29] zlibbioc_1.50.0 rvest_1.0.4
#> [31] GenomicRanges_1.56.1 purrr_1.0.2
#> [33] AnnotationFilter_1.28.0 ggraph_2.2.1
#> [35] RCurl_1.98-1.16 tweenr_2.0.3
#> [37] circlize_0.4.16 GenomeInfoDbData_1.2.12
#> [39] IRanges_2.38.1 ggrepel_0.9.5
#> [41] listenv_0.9.1 parallelly_1.38.0
#> [43] ncdf4_1.23 codetools_0.2-20
#> [45] DelayedArray_0.30.1 DT_0.33
#> [47] xml2_1.3.6 ggforce_0.4.2
#> [49] tidyselect_1.2.1 shape_1.4.6.1
#> [51] UCSC.utils_1.0.0 farver_2.1.2
#> [53] viridis_0.6.5 matrixStats_1.3.0
#> [55] base64enc_0.1-3 jsonlite_1.8.8
#> [57] GetoptLong_1.0.5 tidygraph_1.3.1
#> [59] iterators_1.0.14 foreach_1.5.2
#> [61] tools_4.4.1 progress_1.2.3
#> [63] stringdist_0.9.12 glue_1.7.0
#> [65] gridExtra_2.3 SparseArray_1.4.8
#> [67] xfun_0.47 here_1.0.1
#> [69] MatrixGenerics_1.16.0 GenomeInfoDb_1.40.1
#> [71] withr_3.0.1 BiocManager_1.30.25
#> [73] fastmap_1.2.0 fansi_1.0.6
#> [75] blogdown_1.19 digest_0.6.37
#> [77] R6_2.5.1 colorspace_2.1-1
#> [79] rsvg_2.6.1 utf8_1.2.4
#> [81] tidyr_1.3.1 generics_0.1.3
#> [83] data.table_1.16.0 prettyunits_1.2.0
#> [85] graphlayouts_1.1.1 PSMatch_1.8.0
#> [87] httr_1.4.7 htmlwidgets_1.6.4
#> [89] S4Arrays_1.4.1 pkgconfig_2.0.3
#> [91] gtable_0.3.5 ComplexHeatmap_2.20.0
#> [93] impute_1.78.0 XVector_0.44.0
#> [95] furrr_0.3.1 htmltools_0.5.8.1
#> [97] bookdown_0.40 MALDIquant_1.22.3
#> [99] clue_0.3-65 scales_1.3.0
#> [101] png_0.1-8 knitr_1.48
#> [103] rstudioapi_0.16.0 tzdb_0.4.0
#> [105] reshape2_1.4.4 rjson_0.2.22
#> [107] curl_5.2.2 cachem_1.1.0
#> [109] GlobalOptions_0.1.2 stringr_1.5.1
#> [111] parallel_4.4.1 mzID_1.42.0
#> [113] vsn_3.72.0 pillar_1.9.0
#> [115] grid_4.4.1 vctrs_0.6.5
#> [117] pcaMethods_1.96.0 cluster_2.1.6
#> [119] evaluate_0.24.0 readr_2.1.5
#> [121] cli_3.6.3 compiler_4.4.1
#> [123] rlang_1.1.4 crayon_1.5.3
#> [125] Rdisop_1.64.0 QFeatures_1.14.2
#> [127] ChemmineR_3.56.0 affy_1.82.0
#> [129] plyr_1.8.9 stringi_1.8.4
#> [131] viridisLite_0.4.2 BiocParallel_1.38.0
#> [133] munsell_0.5.1 Biostrings_2.72.1
#> [135] lazyeval_0.2.2 Matrix_1.7-0
#> [137] hms_1.1.3 bit64_4.0.5
#> [139] future_1.34.0 KEGGREST_1.44.1
#> [141] statmod_1.5.0 SummarizedExperiment_1.34.0
#> [143] igraph_2.0.3 memoise_2.0.1
#> [145] affyio_1.74.0 bslib_0.8.0
#> [147] bit_4.0.5 readxl_1.4.3