Here, you can learn how to use tidyMass to do the data processing and analysis for the LC-MS metabolomics data. It contains several parts.

If you are new to R or the tidyverse
We recommend that you start by learning some basics about R and the tidyverse first, then return here when you feel ready. Here are some resources to start learning:
-
Finding Your Way To R, from the RStudio Education team.
-
Learn the tidyverse, from the tidyverse team.
Part 1. Install tidymass
You can learn how to install tidymass, and update it. You can also find here how to download the docker version of tidymass and build your own docker image based on tidymass.
Part 2. massdatabase package and mass_dataset class
You can find here how to download the demo data and create mass_dataset class by yourself. And how to use mass_dataset class organize your omics data and process it.
Part 3. Metabolite annotation
All the metabolite annotation can be found here. You can also learn here how to construct the databases for metid using the massdatabase package.
Part 4. Whole workflow using tidymass
Here, you can learn how to use tidymass for data processing and analysis, from data converting to biological function mining.
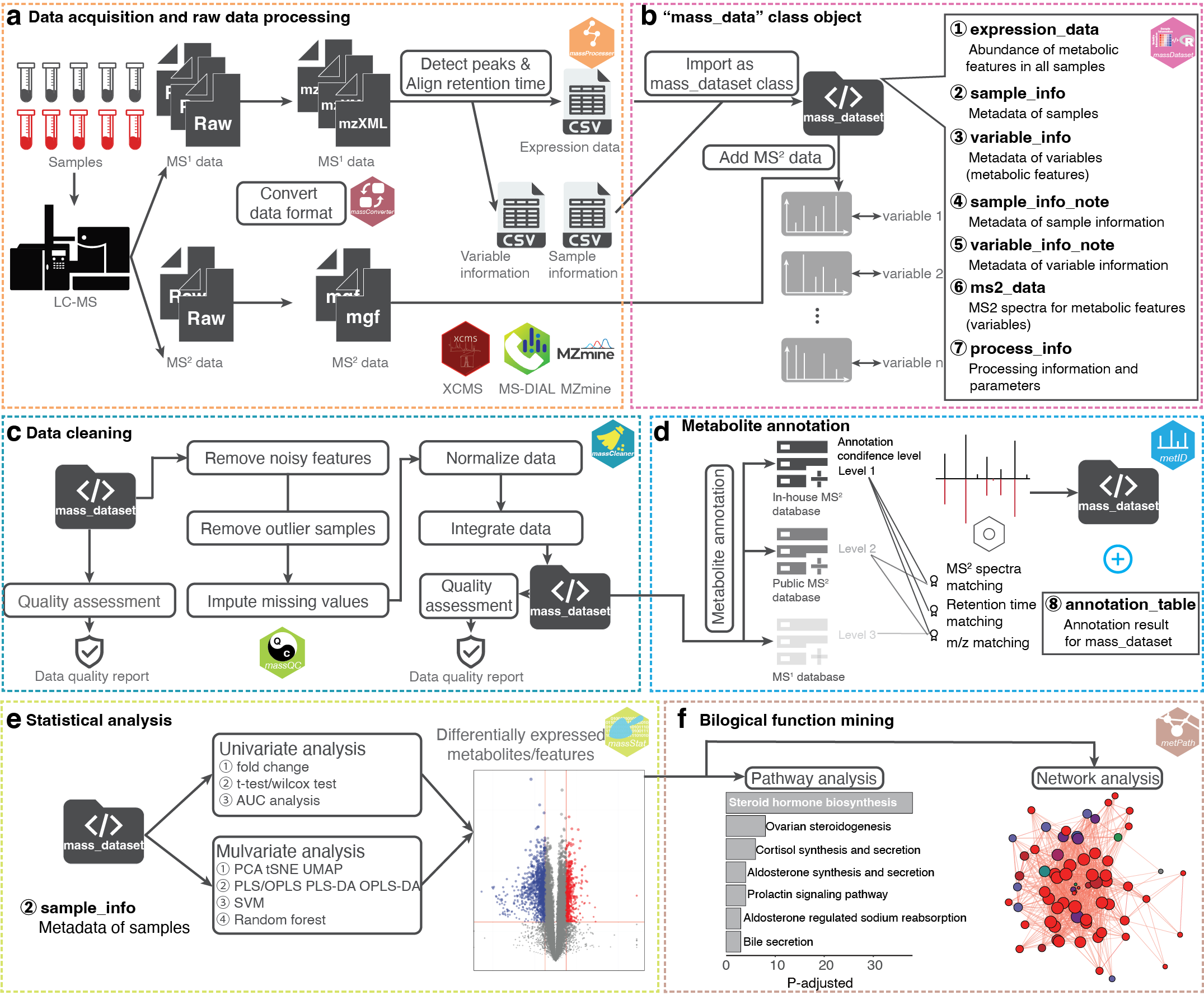
The code, data and docker image of case study in our manuscript are provided here.