Here, you can learn how to use tidyMass to do the data processing and analysis for the LC-MS metabolomics data. It contains several parts.
在这里,您可以学习如何使用 tidyMass 对LC-MS代谢组学数据进行数据处理和分析。它包含几个部分。

If you are new to R or the tidyverse
We recommend that you start by learning some basics about R and the tidyverse first, then return here when you feel ready. Here are some resources to start learning:
我们建议您先学习一些关于R和tidyverse的基础知识,然后在感觉准备好时再回到这里。以下是一些开始学习的资源:
-
Finding Your Way To R, from the RStudio Education team.
-
Learn the tidyverse, from the tidyverse team.
Part 1. Install tidymass
You can learn how to install tidymass, and update it. You can also find here how to download the docker version of tidymass and build your own docker image based on tidymass.
您可以学习如何安装 tidymass,以及如何更新它。您还可以在这里找到如何下载 tidymass 的 Docker 版本,并基于 tidymass 构建自己的 Docker 镜像。
Part 2. massdatabase package and mass_dataset class
You can find here how to download the demo data and create mass_dataset class by yourself. And how to use mass_dataset class organize your omics data and process it.
您可以在这里找到如何下载演示数据并自己创建 mass_dataset 类。以及如何使用 mass_dataset 类来组织您的组学数据并进行处理。
Part 3. Metabolite annotation
All the metabolite annotation can be found here. You can also learn here how to construct the databases for metid using the massdatabase package.
在这里可以找到所有的代谢物注释。您还可以在这里学习如何使用 massdatabase 包构建 metid 的数据库。
Part 4. Whole workflow using tidymass
Here, you can learn how to use tidymass for data processing and analysis, from data converting to biological function mining.
在这里,您可以学习如何使用 tidymass 进行数据处理和分析,从数据转换到生物功能挖掘。
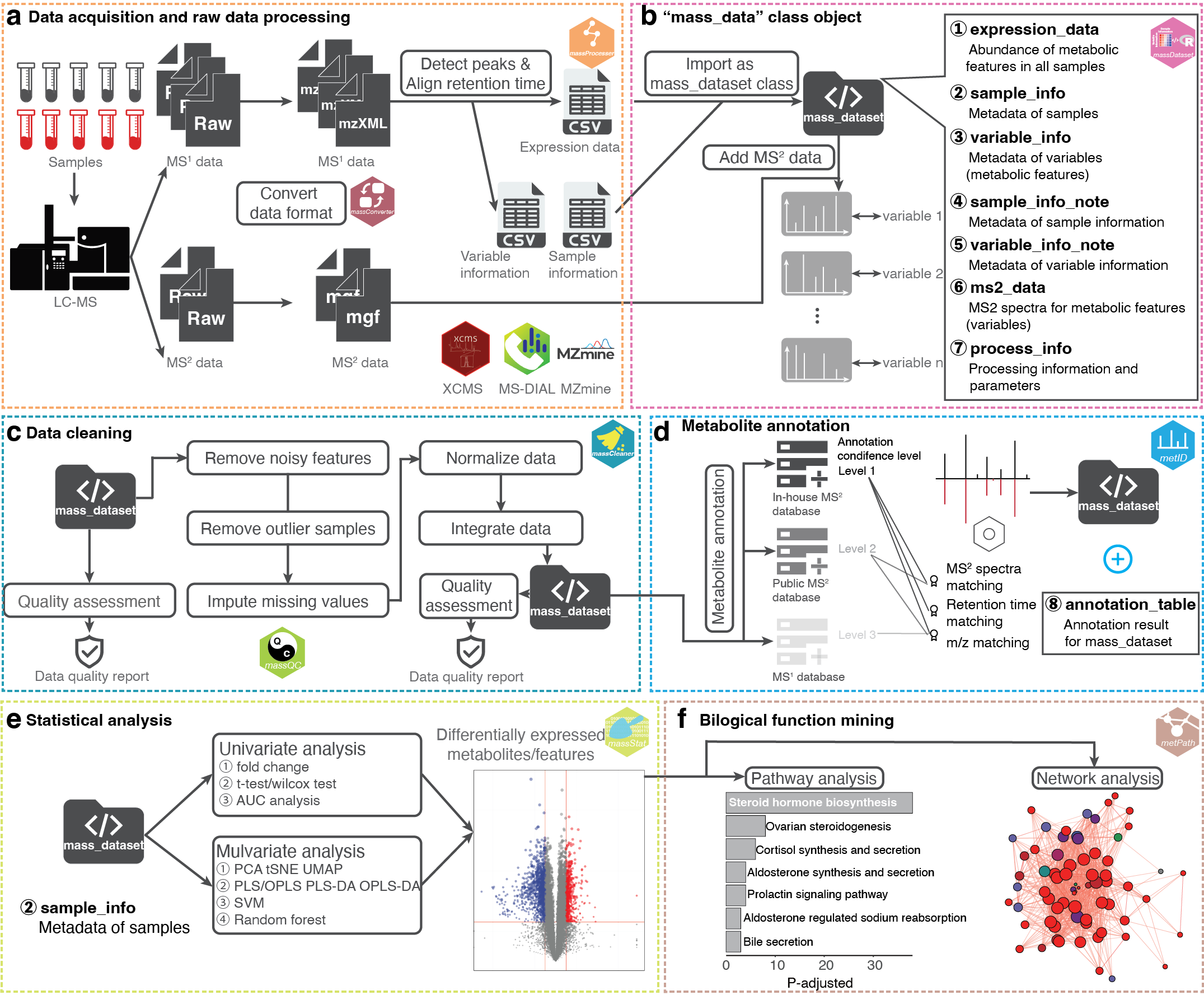
The code, data and docker image of case study in our manuscript are provided here.
在我们的论文中提供了案例研究的代码、数据和Docker镜像。