Data preparation
Download the demo data and refer this article.
We have positive and negative mode. For each mode, we have control, case and QC groups. Control group have 110 samples, and case group have 110 samples as well.
Positive mode
massprocesser package is used to do the raw data processing. Please refer this website.
Code
The code used to do raw data processing.
library(tidymass)
#> Registered S3 method overwritten by 'Hmisc':
#> method from
#> vcov.default fit.models
#> ── Attaching packages ────────────────────────────── tidymass 1.0.9 ──
#> ✔ massdataset 1.0.34 ✔ metid 1.2.33
#> ✔ massprocesser 1.0.10 ✔ masstools 1.0.13
#> ✔ masscleaner 1.0.12 ✔ dplyr 1.1.4
#> ✔ massqc 1.0.7 ✔ ggplot2 3.5.1
#> ✔ massstat 1.0.6 ✔ magrittr 2.0.3
#> ✔ metpath 1.0.8
process_data(
path = "mzxml_ms1_data/POS",
polarity = "positive",
ppm = 10,
peakwidth = c(10, 60),
threads = 4,
output_tic = FALSE,
output_bpc = FALSE,
output_rt_correction_plot = FALSE,
min_fraction = 0.5,
group_for_figure = "QC"
)
Results
All the results will be placed in the folder mzxml_ms1_data/POS/Result. More information can be found here.
You can just load the object, which is a mass_dataset class object.
load("mzxml_ms1_data/POS/Result/object")
object
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 10149 x 259 data.frame]
#> 2.sample_info:[ 259 x 4 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 10149 x 3 data.frame]
#> 10149 variables:M70T73_POS M70T53_POS M70T183_POS ... M923T55_POS M992T641_POS
#> 4.sample_info_note:[ 4 x 2 data.frame]
#> 5.variable_info_note:[ 3 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 2 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:37:06
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:36:42
We can see that there are 10,149 metabolic features in positive mode.
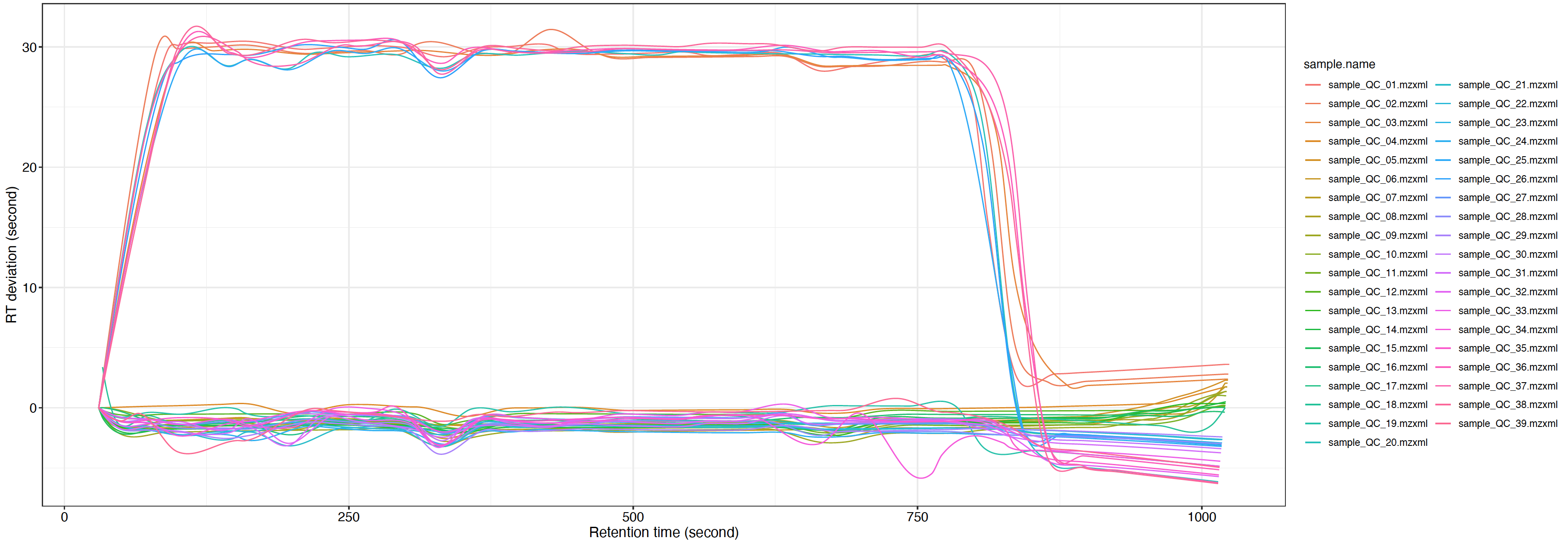
You can use the plot_adjusted_rt() function to get the interactive plot.
load("mzxml_ms1_data/POS/Result/intermediate_data/xdata2")
##set the group_for_figure if you want to show specific groups. And set it as "all" if you want to show all samples.
plot =
massprocesser::plot_adjusted_rt(object = xdata2,
group_for_figure = "QC",
interactive = TRUE)
plot
Negative mode
The processing of negative mode is same with positive mode data.
Code
Same with positive mode, change polarity to negative.
massprocesser::process_data(
path = "mzxml_ms1_data/NEG",
polarity = "negative",
ppm = 10,
peakwidth = c(10, 60),
threads = 4,
output_tic = FALSE,
output_bpc = FALSE,
output_rt_correction_plot = FALSE,
min_fraction = 0.5,
group_for_figure = "QC"
)
Results
Same with positive mode.
load("mzxml_ms1_data/NEG/Result/object")
object
#> --------------------
#> massdataset version: 0.99.8
#> --------------------
#> 1.expression_data:[ 8804 x 259 data.frame]
#> 2.sample_info:[ 259 x 4 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 8804 x 3 data.frame]
#> 8804 variables:M70T712_NEG M70T527_NEG M70T587_NEG ... M941T65_NEG M948T641_NEG
#> 4.sample_info_note:[ 4 x 2 data.frame]
#> 5.variable_info_note:[ 3 x 2 data.frame]
#> 6.ms2_data:[ 0 variables x 0 MS2 spectra]
#> --------------------
#> Processing information
#> 2 processings in total
#> create_mass_dataset ----------
#> Package Function.used Time
#> 1 massdataset create_mass_dataset() 2022-02-23 08:38:19
#> process_data ----------
#> Package Function.used Time
#> 1 massprocesser process_data 2022-02-23 08:38:02
We can see that there are 8,804 metabolic features in negative mode.
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.0
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] metid_1.2.33 metpath_1.0.8 ComplexHeatmap_2.20.0
#> [4] mixOmics_6.28.0 lattice_0.22-6 MASS_7.3-61
#> [7] massstat_1.0.6 tidyr_1.3.1 ggfortify_0.4.17
#> [10] massqc_1.0.7 masscleaner_1.0.12 MSnbase_2.30.1
#> [13] ProtGenerics_1.36.0 S4Vectors_0.42.1 Biobase_2.64.0
#> [16] BiocGenerics_0.50.0 mzR_2.38.0 Rcpp_1.0.13
#> [19] xcms_4.2.3 BiocParallel_1.38.0 massprocesser_1.0.10
#> [22] ggplot2_3.5.1 dplyr_1.1.4 magrittr_2.0.3
#> [25] masstools_1.0.13 massdataset_1.0.34 tidymass_1.0.9
#>
#> loaded via a namespace (and not attached):
#> [1] fs_1.6.4 matrixStats_1.3.0
#> [3] bitops_1.0-8 fit.models_0.64
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 tools_4.4.1
#> [9] doRNG_1.8.6 backports_1.5.0
#> [11] utf8_1.2.4 R6_2.5.1
#> [13] lazyeval_0.2.2 GetoptLong_1.0.5
#> [15] withr_3.0.1 prettyunits_1.2.0
#> [17] gridExtra_2.3 preprocessCore_1.66.0
#> [19] cli_3.6.3 fastDummies_1.7.4
#> [21] sass_0.4.9 mvtnorm_1.3-1
#> [23] robustbase_0.99-4 readr_2.1.5
#> [25] randomForest_4.7-1.1 proxy_0.4-27
#> [27] pbapply_1.7-2 systemfonts_1.1.0
#> [29] foreign_0.8-87 svglite_2.1.3
#> [31] rrcov_1.7-6 MetaboCoreUtils_1.12.0
#> [33] parallelly_1.38.0 itertools_0.1-3
#> [35] limma_3.60.4 readxl_1.4.3
#> [37] rstudioapi_0.16.0 impute_1.78.0
#> [39] generics_0.1.3 shape_1.4.6.1
#> [41] zip_2.3.1 Matrix_1.7-0
#> [43] MALDIquant_1.22.3 fansi_1.0.6
#> [45] abind_1.4-5 lifecycle_1.0.4
#> [47] yaml_2.3.10 SummarizedExperiment_1.34.0
#> [49] SparseArray_1.4.8 crayon_1.5.3
#> [51] PSMatch_1.8.0 KEGGREST_1.44.1
#> [53] pillar_1.9.0 knitr_1.48
#> [55] GenomicRanges_1.56.1 rjson_0.2.22
#> [57] corpcor_1.6.10 codetools_0.2-20
#> [59] glue_1.7.0 pcaMethods_1.96.0
#> [61] data.table_1.16.0 remotes_2.5.0
#> [63] MultiAssayExperiment_1.30.3 vctrs_0.6.5
#> [65] png_0.1-8 cellranger_1.1.0
#> [67] gtable_0.3.5 cachem_1.1.0
#> [69] xfun_0.47 openxlsx_4.2.7
#> [71] S4Arrays_1.4.1 tidygraph_1.3.1
#> [73] pcaPP_2.0-5 ncdf4_1.23
#> [75] iterators_1.0.14 statmod_1.5.0
#> [77] robust_0.7-5 progress_1.2.3
#> [79] GenomeInfoDb_1.40.1 rprojroot_2.0.4
#> [81] bslib_0.8.0 affyio_1.74.0
#> [83] rpart_4.1.23 colorspace_2.1-1
#> [85] DBI_1.2.3 Hmisc_5.1-3
#> [87] nnet_7.3-19 tidyselect_1.2.1
#> [89] compiler_4.4.1 MassSpecWavelet_1.70.0
#> [91] htmlTable_2.4.3 DelayedArray_0.30.1
#> [93] plotly_4.10.4 bookdown_0.40
#> [95] checkmate_2.3.2 scales_1.3.0
#> [97] DEoptimR_1.1-3 affy_1.82.0
#> [99] stringr_1.5.1 digest_0.6.37
#> [101] rmarkdown_2.28 XVector_0.44.0
#> [103] htmltools_0.5.8.1 pkgconfig_2.0.3
#> [105] base64enc_0.1-3 MatrixGenerics_1.16.0
#> [107] fastmap_1.2.0 rlang_1.1.4
#> [109] GlobalOptions_0.1.2 htmlwidgets_1.6.4
#> [111] UCSC.utils_1.0.0 farver_2.1.2
#> [113] jquerylib_0.1.4 jsonlite_1.8.8
#> [115] MsExperiment_1.6.0 mzID_1.42.0
#> [117] RCurl_1.98-1.16 Formula_1.2-5
#> [119] GenomeInfoDbData_1.2.12 patchwork_1.2.0
#> [121] munsell_0.5.1 viridis_0.6.5
#> [123] MsCoreUtils_1.16.1 vsn_3.72.0
#> [125] furrr_0.3.1 stringi_1.8.4
#> [127] ggraph_2.2.1 zlibbioc_1.50.0
#> [129] plyr_1.8.9 parallel_4.4.1
#> [131] listenv_0.9.1 ggrepel_0.9.5
#> [133] Biostrings_2.72.1 MsFeatures_1.12.0
#> [135] graphlayouts_1.1.1 hms_1.1.3
#> [137] Spectra_1.14.1 circlize_0.4.16
#> [139] igraph_2.0.3 QFeatures_1.14.2
#> [141] rngtools_1.5.2 reshape2_1.4.4
#> [143] XML_3.99-0.17 evaluate_0.24.0
#> [145] blogdown_1.19 BiocManager_1.30.25
#> [147] tzdb_0.4.0 foreach_1.5.2
#> [149] missForest_1.5 tweenr_2.0.3
#> [151] purrr_1.0.2 polyclip_1.10-7
#> [153] future_1.34.0 clue_0.3-65
#> [155] ggforce_0.4.2 AnnotationFilter_1.28.0
#> [157] e1071_1.7-14 RSpectra_0.16-2
#> [159] ggcorrplot_0.1.4.1 viridisLite_0.4.2
#> [161] class_7.3-22 rARPACK_0.11-0
#> [163] tibble_3.2.1 memoise_2.0.1
#> [165] ellipse_0.5.0 IRanges_2.38.1
#> [167] cluster_2.1.6 globals_0.16.3
#> [169] here_1.0.1