Other tools in metflow2
Xiaotao Shen PhD (https://www.shenxt.info/)
true
Created on 2020-04-01 and updated on 2021-04-07
Source:vignettes/other_tools.Rmd
other_tools.Rmdmetflow2 also provide some other useful tools.
Metabolite ID convert
Note: This function has been deprecated, try to new function in
tinyToolspackage.
transID() function can be used to convert metabolites in more than 225 databases. It is based on the http://cts.fiehnlab.ucdavis.edu/ and https://www.chemspider.com/InChI.asmx.
Convert chemical ID based on http://cts.fiehnlab.ucdavis.edu/
You can use the databaseName() function to get the databases supported in transID().
library(metflow2)
#> ✓ xcms 3.12.0 ✓ MSnbase 2.16.0
#> ✓ mzR 2.24.1
library(tidyverse)
#> ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.0 ──
#> ✓ ggplot2 3.3.3 ✓ purrr 0.3.4
#> ✓ tibble 3.1.0 ✓ dplyr 1.0.4
#> ✓ tidyr 1.1.3 ✓ stringr 1.4.0
#> ✓ readr 1.4.0 ✓ forcats 0.5.0
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> x dplyr::collect() masks xcms::collect()
#> x dplyr::combine() masks MSnbase::combine(), Biobase::combine(), BiocGenerics::combine()
#> x tidyr::expand() masks S4Vectors::expand()
#> x dplyr::filter() masks stats::filter()
#> x dplyr::first() masks S4Vectors::first()
#> x dplyr::groups() masks xcms::groups()
#> x dplyr::lag() masks stats::lag()
#> x ggplot2::Position() masks BiocGenerics::Position(), base::Position()
#> x purrr::reduce() masks MSnbase::reduce()
#> x dplyr::rename() masks S4Vectors::rename()
database <- metflow2::databaseName(server = "http://cts.fiehnlab.ucdavis.edu/service/convert")
#> databaseName() is deprecated, please use the trans_id_database() in tinyTools.
#> 225 databases are supported in server http://cts.fiehnlab.ucdavis.edu/service/convert for 'from'.
#> 226 databases are supported in server http://cts.fiehnlab.ucdavis.edu/service/convert for 'to'.
database$From
#> # A tibble: 225 x 1
#> From
#> <chr>
#> 1 AAA Chemistry
#> 2 ABBLIS Chemicals
#> 3 Abbott Labs
#> 4 ABI Chem
#> 5 AbMole Bioscience
#> 6 Acesobio
#> 7 Achemica
#> 8 Acorn PharmaTech
#> 9 Active Biopharma
#> 10 Adooq BioScience
#> # … with 215 more rows
database$To
#> # A tibble: 226 x 1
#> From
#> <chr>
#> 1 AAA Chemistry
#> 2 ABBLIS Chemicals
#> 3 Abbott Labs
#> 4 ABI Chem
#> 5 AbMole Bioscience
#> 6 Acesobio
#> 7 Achemica
#> 8 Acorn PharmaTech
#> 9 Active Biopharma
#> 10 Adooq BioScience
#> # … with 216 more rowsThen you can convert ID using transID() function.
transID(query = "C00001", from = "KEGG",
to = "PubChem SID", top = 2,
server = "http://cts.fiehnlab.ucdavis.edu/service/convert")
#> transID() is deprecated, please use the trans_ID() in tinyTools.
#> # A tibble: 2 x 2
#> KEGG `PubChem SID`
#> <chr> <chr>
#> 1 C00001 103059594
#> 2 C00001 10322299The from and to argument must be from databaseName() function.
transID(query = "C00001", from = "KEGG",
to = "Human Metabolome Database", top = 2,
server = "http://cts.fiehnlab.ucdavis.edu/service/convert")
#> # A tibble: 1 x 2
#> KEGG `Human Metabolome Database`
#> <chr> <chr>
#> 1 C00001 HMDB0002111Convert chemical ID based on https://www.chemspider.com/InChI.asmx
Another databsae https://www.chemspider.com/InChI.asmx also support ID conveter. First please use databaseName() to know which databases are supported in this website.
database <- databaseName(server = "https://www.chemspider.com/InChI.asmx")
#> 9 are supported in server https://www.chemspider.com/InChI.asmx.
database
#> # A tibble: 9 x 2
#> From To
#> <chr> <chr>
#> 1 csid mol
#> 2 inchikey csid
#> 3 inchikey inchi
#> 4 inchikey mol
#> 5 inchi csid
#> 6 inchi inchikey
#> 7 inchi mol
#> 8 inchi smiles
#> 9 smiles inchiHere we first use the http://cts.fiehnlab.ucdavis.edu/service/convert convert C00001 to get it’s InChIKey ID.
transID(query = "C00010",
from = "KEGG",
to = "InChIKey",
top = 1,
server = "http://cts.fiehnlab.ucdavis.edu/service/convert")
#> # A tibble: 1 x 2
#> KEGG InChIKey
#> <chr> <chr>
#> 1 C00010 RGJOEKWQDUBAIZ-IBOSZNHHSA-NThen we use InChIKey ID to get its smiles.
inchi <-
transID(query = "RGJOEKWQDUBAIZ-IBOSZNHHSA-N",
from = "inchikey",
to = "inchi",
top = 2,
server = "https://www.chemspider.com/InChI.asmx")
inchi
#> [1] "InChI=1S/C21H36N7O16P3S/c1-21(2,16(31)19(32)24-4-3-12(29)23-5-6-48)8-41-47(38,39)44-46(36,37)40-7-11-15(43-45(33,34)35)14(30)20(42-11)28-10-27-13-17(22)25-9-26-18(13)28/h9-11,14-16,20,30-31,48H,3-8H2,1-2H3,(H,23,29)(H,24,32)(H,36,37)(H,38,39)(H2,22,25,26)(H2,33,34,35)/t11-,14-,15-,16+,20-/m1/s1"
smiles <-
transID(query = inchi,
from = "inchi",
to = "smiles",
top = 2,
server = "https://www.chemspider.com/InChI.asmx")
smiles
#> [1] "CC(C)(COP(=O)(O)OP(=O)(O)OC[C@@H]1[C@H]([C@H]([C@H](n2cnc3c(N)ncnc23)O1)O)OP(=O)(O)O)[C@H](C(=NCCC(=NCCS)O)O)O"Get metabolite class
Note: This function has been deprecated, try to new function in
tinyToolspackage.
We can use get_metclass() function to get the metabolite class based on classyfire.
We need the inchikey for get_metclass() function, so if we don’t have the inchikey, we can use the transID() function to get their inchikey.
inchikey <-
transID(query = "C00001",
from = "KEGG",
to = "InChIKey",
top = 1,
server = "http://cts.fiehnlab.ucdavis.edu/service/convert")
#> transID() is deprecated, please use the trans_ID() in tinyTools.
inchikey <- inchikey$InChIKey
inchikey
#> [1] "XLYOFNOQVPJJNP-UHFFFAOYSA-N"
class <- get_metclass(inchikey = inchikey)
#> get_metclass() is deprecated, please use the get_compound_class() in tinyTools.class is a classfire class.
class
#> ── classyfire Object ──────────────────────────────────────── metflow2 v0.9.2 ──
#> Object Size: 8.6 Kb
#>
#> Information:
#> SMILES: O
#> InChIKey: InChIKey=XLYOFNOQVPJJNP-UHFFFAOYSA-N
#> Formula: H2O
#> Mass: 18.015
#> Kingdom : Inorganic compounds
#> └─Superclass : Homogeneous non-metal compounds
#> └─Class : Homogeneous other non-metal compounds
class@compound_info
#> # A tibble: 4 x 2
#> name value
#> <chr> <chr>
#> 1 SMILES O
#> 2 InChIKey InChIKey=XLYOFNOQVPJJNP-UHFFFAOYSA-N
#> 3 Formula H2O
#> 4 Mass 18.015
class@taxonomy_tree
#> # A tibble: 3 x 2
#> name value
#> <chr> <chr>
#> 1 Kingdom Inorganic compounds
#> 2 Superclass Homogeneous non-metal compounds
#> 3 Class Homogeneous other non-metal compounds
class@classification_info
#> # A tibble: 9 x 2
#> name value
#> <chr> <chr>
#> 1 Kingdom Inorganic compounds
#> 2 Superclass Homogeneous non-metal compounds
#> 3 Class Homogeneous other non-metal compounds
#> 4 Subclass Not available
#> 5 Intermediate Tree Nodes Not available
#> 6 Direct Parent Homogeneous other non-metal compounds
#> 7 Alternative Parents Not available
#> 8 Molecular Framework Not available
#> 9 Substituents Homogeneous other non metal
class@description
#> # A tibble: 1 x 2
#> name value
#> <chr> <chr>
#> 1 Description This compound belongs to the class of inorganic compounds known a…
class@external_descriptors
#> # A tibble: 1 x 2
#> name value
#> <chr> <chr>
#> 1 External Descriptors CHEBI:15377 :Generate running list
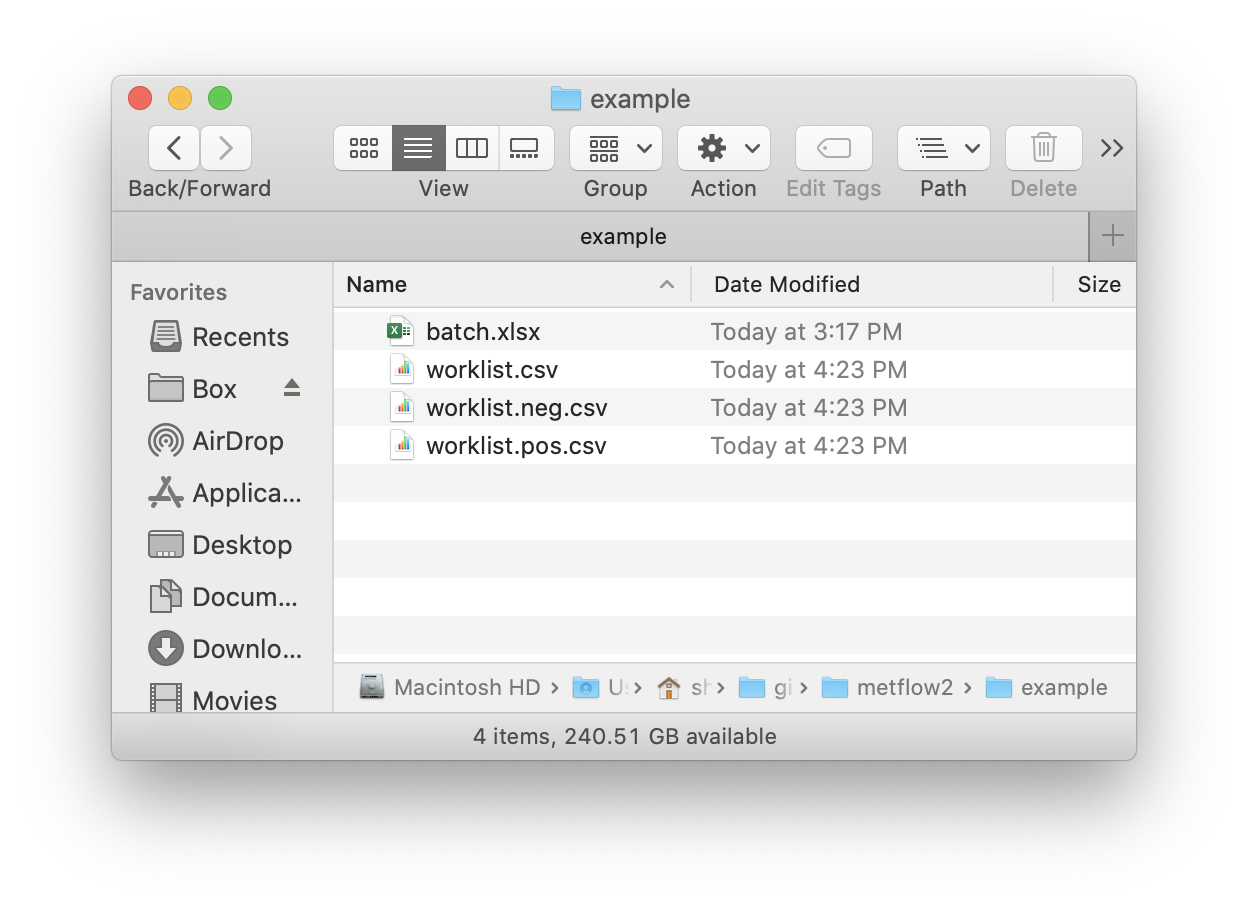
We can use the getWorklist() function in metflow2 to generate a working list.
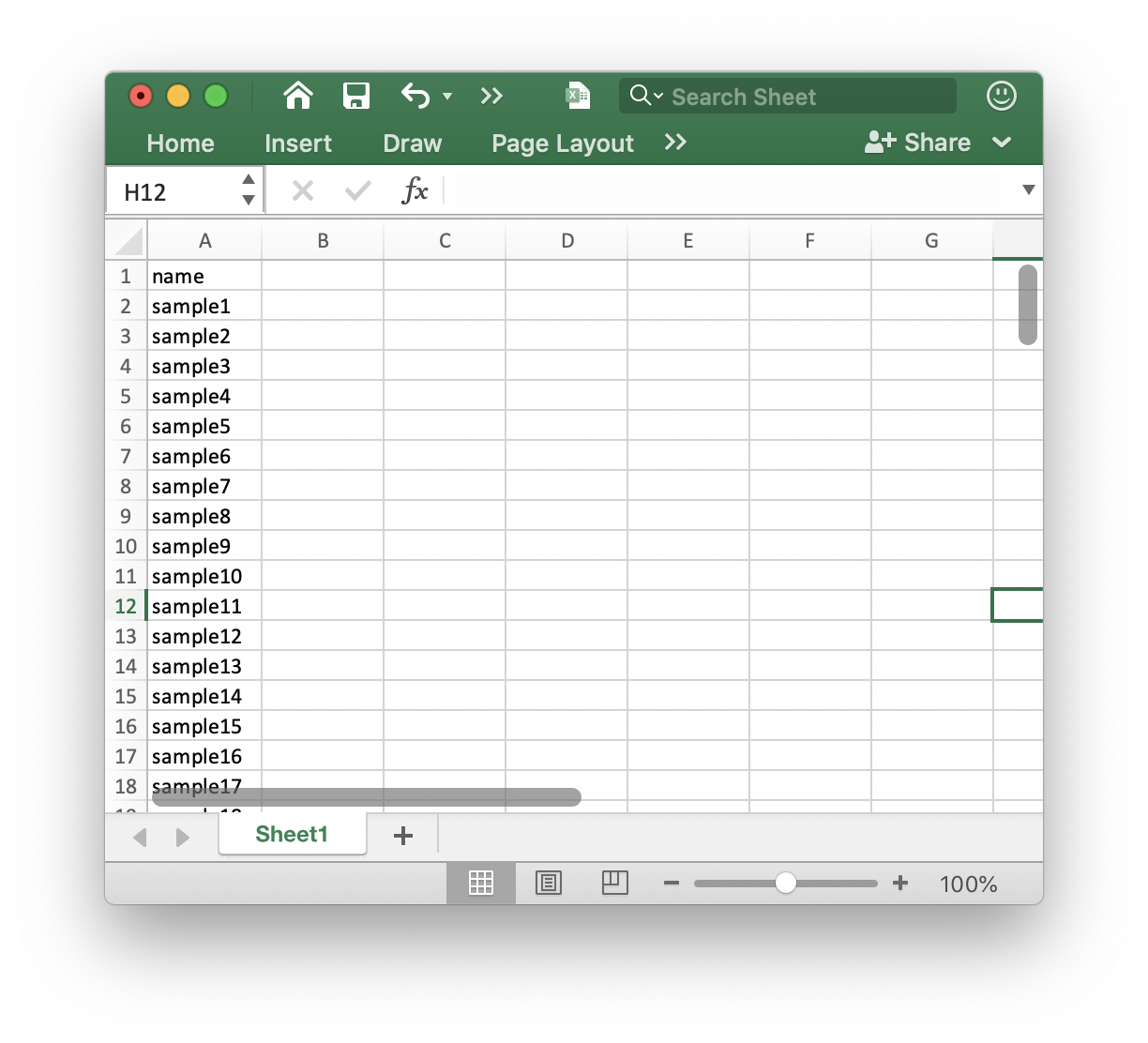
First, we should creat a batch.xlsx and put it in a folder. We use the demo data in demoData package.
The batch.xlsx is like this:
##creat a folder nameed as example
path <- file.path(".", "example")
dir.create(path = path, showWarnings = FALSE)
##get demo data
demo_data <- system.file("metflow2", package = "demoData")
file.copy(from = file.path(demo_data, "batch.xlsx"),
to = path, overwrite = TRUE, recursive = TRUE)
#> [1] TRUENext, run the getWorklist() function.
getWorklist(
table.name = "batch.xlsx",
instrument = c("Thermo", "Agilent", "AB"),
each.mode.number = 32,
randommethod = c("no", "position", "injection"),
samplenumber = NULL,
QCstep = 8,
conditionQCnumber = 8,
qc.index.from = 1,
dir = "D:\\Liang\\data\\PS4U\\HILIC\\batch3\\",
method.path = "D:\\Liang\\Method\\urine\\HILIC\\",
ms1.method.pos = "ZIC-HILIC_MS_pos",
ms1.method.neg = "ZIC-HILIC_MS_neg",
ms2.method.pos = c(
"ZIC-HILIC_MSMS_pos_NCE25",
"ZIC-HILIC_MSMS_pos_NCE25",
"ZIC-HILIC_MSMS_pos_NCE25",
"ZIC-HILIC_MSMS_pos_NCE25"
),
ms2.method.neg = c(
"ZIC-HILIC_MSMS_neg_NCE25",
"ZIC-HILIC_MSMS_neg_NCE25",
"ZIC-HILIC_MSMS_neg_NCE25",
"ZIC-HILIC_MSMS_neg_NCE25"
),
path = path
)Then, the worklists will generated in this folder.