Missing value imputation
Xiaotao Shen (https://www.shenxt.info/)
true
Created on 2020-04-01 and updated on 2021-04-07
Source:vignettes/missing_value_imputation.Rmd
missing_value_imputation.RmdWe can use metflow2 for missing value (MV) imputation.
First, we need to prepare samples for metflow2.
Data preparation
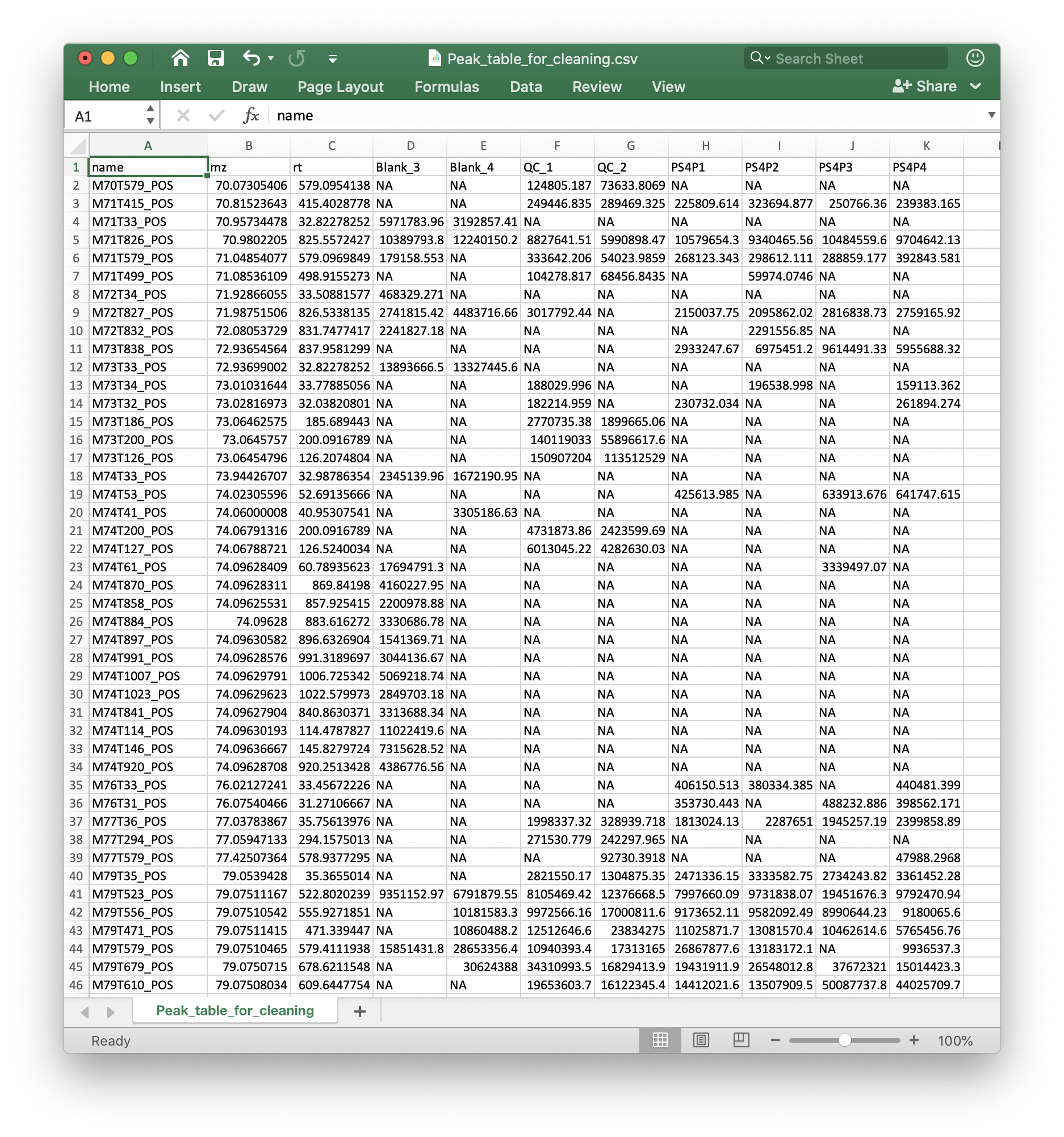
Peak table
The peak table (csv format) can be from any software. We recommend that you use the Peak_table_for_cleaning.csv from processData() function from metflow2.
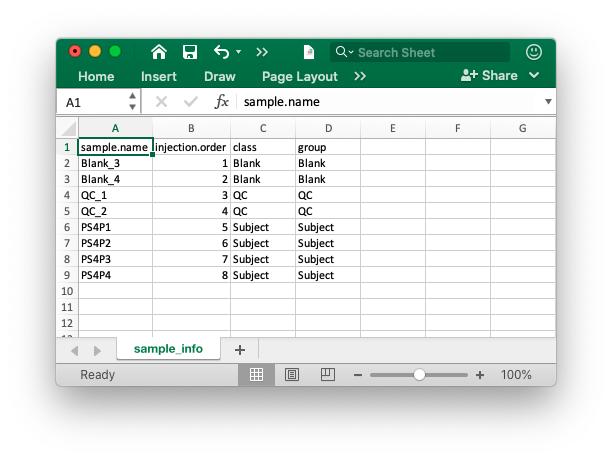
If you use other software, please make sure that the top 3 columns are name (peak name), mz and rt (retention time, second). And the left column are sample intensity.
Read data
Then place the peak table and sample information in a folder. We use the demo data from demoData package.
Load demo data
##creat a folder nameed as example
path <- file.path(".", "example")
dir.create(path = path, showWarnings = FALSE)
##get demo data
demo_data <- system.file("metflow2", package = "demoData")
file.copy(from = file.path(demo_data, dir(demo_data)),
to = path, overwrite = TRUE, recursive = TRUE)
#> [1] TRUE TRUE TRUE TRUE TRUEHere, we have two peak tables, batch1.data.csv and batch2.data.csv, and sample_info.csv are in your ./example folder.
Creat metflowClass object
object <-
create_metflow_object(
ms1.data = c("batch1.data.csv", "batch2.data.csv"),
sample.information = "sample_info.csv",
path = path
)
#> `creatMetflowObject()` is deprecated, please use `create_metflow_object()`Reading data...
#> --------------------------------------------------------------
#> --------------------------------------------------------------
#> --------------------------------------------------------------
#> Summary:
#> Check result OK Warning Error
#> batch1 Valid 3 0 0
#> batch2 Valid 3 0 0
#> sample.info Valid 9 0 0
#>
#>
#> data:
#> Batch 1 is valid.
#> Batch 2 is valid.
#>
#> sample.info:
#> sample.info is valid.object is a metflowClass object, so you can print it in the console.
object
#> --------------------
#> metflow2 version: 0.1.0
#> --------------------
#> MS1 data
#> There are 2 peak tables in your MS1 data.
#> Peak.number Column.number
#> Batch1 500 220
#> Batch2 500 220
#> --------------------
#> There are 434 samples in your MS1 data.
#> Class Number
#> 1 QC 50
#> 2 Subject 384
#> --------------------
#> Group Number
#> 1 0 103
#> 2 1 281
#> 3 QC 50
#> --------------------
#> Processing
#> There are no processing for your data.Align different batches
Because there are two batch peak tables, so first we must align them.
object <- align_batch(
object = object,
combine.mz.tol = 15,
combine.rt.tol = 30,
use.int.tol = FALSE
)
#> Rough aligning...
#> Accurate aligning...
object
#> --------------------
#> metflow2 version: 0.1.0
#> --------------------
#> MS1 data
#> There are 1 peak tables in your MS1 data.
#> Peak.number Column.number
#> Batch1 354 437
#> --------------------
#> There are 434 samples in your MS1 data.
#> Class Number
#> 1 QC 50
#> 2 Subject 384
#> --------------------
#> Group Number
#> 1 0 103
#> 2 1 281
#> 3 QC 50
#> --------------------
#> Processing
#> alignBatch ----------
#> Parameter Value
#> 1 combine.mz.tol 15
#> 2 combine.rt.tol 30
#> combine.mz.tol
#> combine.rt.tolMissing value processing
First, we should remove some peaks and samples which have a lot of missing values.
Remove noisy peaks and outlier samples
We use filter_peaks() function to filter noisy peaks.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
type = "any",
min.subject.blank.ratio = 2,
according.to = "class",
which.group = "QC"
)
#> QC are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 353 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.There are three creteria in filter_peaks() to remove peaks:
- Missing value
If you want to remove peaks according to peaks NA in samples. You should what groups you want to use. For example, if you want to remove peaks which have more than 50% NA in QC samples, you can set according.to as class, because QC group is defined in class column in sample.info, then the which.group should be set as QC, and min.fraction as 0.5.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
according.to = "class",
which.group = "QC"
)
#> QC are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 353 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.If you want to remove peaks which have more than 50% NAs in QC and/or Subject samples.You can set which.group as c("QC", "Subject"), a vector. And the type is all means the peaks should meet the min.fraction in QC and Subject, and any means that the peaks meet the min.fraction in QC or Subject.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
according.to = "class",
which.group = c("QC", "Subject"),
type = "all"
)
#> QC;Subject are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 330 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
according.to = "class",
which.group = c("QC", "Subject"),
type = "any"
)
#> QC;Subject are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 354 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.- Blank samples
If you have Blank samples in your data, you can also remove some peaks according to Blank samples. If min.subject.blank.ratio is set as 2, it means thay only the peak whoes intensitys in which.group samples is higher than 2 times of intensitys in Blank samples will be left. If min.subject.blank.ratio is set < 1, no peaks will be removed.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
according.to = "class",
which.group = c("QC", "Subject"),
type = "any",
min.subject.blank.ratio = 0
)
#> QC;Subject are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 354 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.
object2 <- filter_peaks(
object = object,
min.fraction = 0.5,
according.to = "class",
which.group = c("QC", "Subject"),
type = "all",
min.subject.blank.ratio = 2
)
#> QC;Subject are in the class .
#> --------------------
#> Removing peaks according to NA in samples...
#> 330 out of 354 peaks are remained.
#> --------------------
#> Removing peaks according to blank samples...
#> No Blanks in your data.
#> --------------------
#> Removing peaks according to QC dilution samples...
#> All done.
object2
#> --------------------
#> metflow2 version: 0.1.0
#> --------------------
#> MS1 data
#> There are 1 peak tables in your MS1 data.
#> Peak.number Column.number
#> Batch1 330 437
#> --------------------
#> There are 434 samples in your MS1 data.
#> Class Number
#> 1 QC 50
#> 2 Subject 384
#> --------------------
#> Group Number
#> 1 0 103
#> 2 1 281
#> 3 QC 50
#> --------------------
#> Processing
#> alignBatch ----------
#> Parameter Value
#> 1 combine.mz.tol 15
#> 2 combine.rt.tol 30
#> filterPeaks ----------
#> Parameter Value
#> 1 min.fraction 0.5
#> 2 which.group QC;Subject
#> combine.mz.tol
#> combine.rt.tol
#> min.fraction
#> which.group
#> 0.5
#> QC;SubjectAfter remove the noisy peaks, there are only 5864 peaks.
Remove outlier samples
Nest, we should remove some samples which have a lot of missing values.
object2 <- filter_samples(object = object2,
min.fraction.peak = 0.5)
#> Samples with MV ratio larger than 0.5 :
#>
#> All done!min.fraction.peak is set as 0.8 means that only the Subject or QC samples with more than 80% peaks are no missing values will be left.
We can get the missing values distribution in samples:
get_mv_plot_samples(object = object2, interactive = TRUE)Missing value imputation
The function impute_mv() is used to do missing value imputation.
object2 <- impute_mv(object = object2,
method = "knn")
object2
#> --------------------
#> metflow2 version: 0.1.0
#> --------------------
#> MS1 data
#> There are 1 peak tables in your MS1 data.
#> Peak.number Column.number
#> Batch1 330 437
#> --------------------
#> There are 434 samples in your MS1 data.
#> Class Number
#> 1 QC 50
#> 2 Subject 384
#> --------------------
#> Group Number
#> 1 0 103
#> 2 1 281
#> 3 QC 50
#> --------------------
#> Processing
#> alignBatch ----------
#> Parameter Value
#> 1 combine.mz.tol 15
#> 2 combine.rt.tol 30
#> filterPeaks ----------
#> Parameter Value
#> 1 min.fraction 0.5
#> 2 which.group QC;Subject
#> filterSample ----------
#> Parameter Value
#> 1 min.fraction.peak 0.5
#> imputeMV ----------
#> Parameter Value
#> 1 method knn
#> 2 k 10
#> 3 rowmax 0.5
#> 4 colmax 0.8
#> 5 maxp 1500
#> 6 rng.seed 362436069
#> 7 maxiter 10
#> 8 ntree 100
#> 9 decreasing FALSE
#> 10 nPcs 2
#> 11 maxSteps 100
#> 12 threshold 1e-04
#> combine.mz.tol
#> combine.rt.tol
#> min.fraction
#> which.group
#> 0.5
#> QC;Subject
#> min.fraction.peak
#> method
#> k
#> rowmax
#> colmax
#> maxp
#> rng.seed
#> maxiter
#> ntree
#> decreasing
#> nPcs
#> maxSteps
#> threshold
#> knn
#> 10
#> 0.5
#> 0.8
#> 1500
#> 362436069
#> 10
#> 100
#> FALSE
#> 2
#> 100
#> 1e-04Note: Only the
SubjectandQCsamples are imputed.
So now, the object2 have been imputed using KNN method.
If you want to output the peak_table and sample_info, you can use get_data() function.
##get the peak table
peak_table2 <- get_data(object = object2, slot = "peak.table")
peak_table2
#> # A tibble: 330 x 437
#> name mz rt QC11 QC12 QC22 QC23 QC24 QC25 QC26 QC27
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 M72T… 72.1 47.1 2.17e6 2.31e6 1.91e6 2.55e6 2.72e6 4.27e6 4.07e6 2.73e6
#> 2 M86T… 86.1 93.8 9.28e6 1.00e7 3.87e6 3.96e6 3.98e6 5.41e6 3.65e6 3.77e6
#> 3 M86T… 86.1 74.6 2.98e6 3.40e6 1.95e6 2.72e6 2.74e6 3.54e6 2.49e6 2.70e6
#> 4 M90T… 89.5 654. 1.09e6 1.46e6 1.45e6 1.41e6 1.40e6 1.56e6 1.44e6 1.16e6
#> 5 M100… 100. 151. 7.78e6 8.39e6 7.28e6 7.94e6 8.34e6 9.00e6 8.64e6 8.47e6
#> 6 M104… 104. 30.9 1.20e7 1.36e7 1.29e7 1.43e7 1.36e7 1.34e7 1.35e7 1.37e7
#> 7 M105… 105. 136. 1.39e6 1.48e6 2.19e5 2.33e5 2.45e5 2.80e5 3.41e5 1.91e5
#> 8 M114… 114. 670. 1.33e5 1.55e5 8.58e5 6.48e5 6.64e5 6.45e5 7.09e5 6.97e5
#> 9 M114… 114. 37.6 1.73e6 1.62e6 1.84e6 2.00e6 1.80e6 2.19e6 1.96e6 2.11e6
#> 10 M116… 116. 33.8 2.15e6 2.14e6 1.35e6 1.56e6 1.49e6 1.93e6 1.84e6 1.51e6
#> # … with 320 more rows, and 426 more variables: QC28 <dbl>, QC29 <dbl>,
#> # QC30 <dbl>, QC31 <dbl>, QC13 <dbl>, QC32 <dbl>, QC33 <dbl>, QC34 <dbl>,
#> # QC35 <dbl>, QC14 <dbl>, QC15 <dbl>, QC16 <dbl>, QC17 <dbl>, QC18 <dbl>,
#> # QC19 <dbl>, QC20 <dbl>, QC21 <dbl>, EC4349 <dbl>, EC7415 <dbl>,
#> # ECFD103 <dbl>, EC3972 <dbl>, EC6120 <dbl>, EC6561 <dbl>, EC7522 <dbl>,
#> # EC7401 <dbl>, EC34A1771 <dbl>, EC6594 <dbl>, EC53A2215 <dbl>,
#> # EC47A2046 <dbl>, EC6557 <dbl>, EC6517 <dbl>, EC7221 <dbl>, EC7560 <dbl>,
#> # EC4365 <dbl>, EC4182 <dbl>, ECA1469 <dbl>, EC4455 <dbl>, EC17A1458 <dbl>,
#> # EC4641 <dbl>, EC6364 <dbl>, EC7460 <dbl>, EC6891 <dbl>, ECA1467 <dbl>,
#> # EC4121 <dbl>, EC6A1215 <dbl>, EC24A1581 <dbl>, EC6349 <dbl>, EC7447 <dbl>,
#> # EC4177 <dbl>, ECFB131 <dbl>, EC26A1605 <dbl>, EC6401 <dbl>, EC7586 <dbl>,
#> # EC6591 <dbl>, EC30A1671 <dbl>, ECA558 <dbl>, EC4370 <dbl>, EC4654 <dbl>,
#> # EC4374 <dbl>, EC4565 <dbl>, EC563 <dbl>, EC6602 <dbl>, EC3751 <dbl>,
#> # ECFB60 <dbl>, EC7631 <dbl>, EC6513 <dbl>, EC4638 <dbl>, EC8365 <dbl>,
#> # EC4149 <dbl>, EC4536 <dbl>, EC4293 <dbl>, ECFB123_1 <dbl>, EC6132 <dbl>,
#> # EC7403 <dbl>, EC4302 <dbl>, EC4385 <dbl>, EC6291 <dbl>, EC6548 <dbl>,
#> # EC6461 <dbl>, EC6742 <dbl>, EC6400 <dbl>, EC7597 <dbl>, EC6748 <dbl>,
#> # EC6101 <dbl>, ECFB123_2 <dbl>, EC6305 <dbl>, EC4106 <dbl>, EC4265 <dbl>,
#> # EC4136 <dbl>, EC4206 <dbl>, EC7372 <dbl>, EC6208 <dbl>, EC4291 <dbl>,
#> # EC6752 <dbl>, EC4482 <dbl>, EC6893 <dbl>, EC7360 <dbl>, EC7590 <dbl>,
#> # EC4781 <dbl>, EC7234 <dbl>, …
##get the sample inforamtion
sample_info2 <- get_data(object = object2, slot = "sample.info")
sample_info2
#> # A tibble: 434 x 5
#> sample.name injection.order class batch group
#> <chr> <dbl> <chr> <dbl> <chr>
#> 1 QC11 1 QC 1 QC
#> 2 EC6225 2 Subject 1 1
#> 3 EC567 3 Subject 1 0
#> 4 EC5A1395 4 Subject 1 1
#> 5 EC4604 5 Subject 1 1
#> 6 EC7542 6 Subject 1 1
#> 7 EC7528 7 Subject 1 1
#> 8 EC6345 8 Subject 1 1
#> 9 EC6108 9 Subject 1 1
#> 10 QC12 10 QC 1 QC
#> # … with 424 more rows