Data preparation
Load the differential expressional metabolites.
library(tidymass)
#> Registered S3 method overwritten by 'Hmisc':
#> method from
#> vcov.default fit.models
#> ── Attaching packages ──────────────────────────────────────── tidymass 1.0.9 ──
#> ✔ massdataset 1.0.34 ✔ metid 1.2.34
#> ✔ massprocesser 1.0.10 ✔ masstools 1.0.13
#> ✔ masscleaner 1.0.12 ✔ dplyr 1.1.4
#> ✔ massqc 1.0.7 ✔ ggplot2 3.5.1
#> ✔ massstat 1.0.6 ✔ magrittr 2.0.3
#> ✔ metpath 1.0.8
library(tidyverse)
#> ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
#> ✔ forcats 1.0.0 ✔ readr 2.1.5
#> ✔ lubridate 1.9.3 ✔ stringr 1.5.1
#> ✔ purrr 1.0.2 ✔ tibble 3.2.1
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> ✖ xcms::collect() masks dplyr::collect()
#> ✖ MSnbase::combine() masks Biobase::combine(), BiocGenerics::combine(), dplyr::combine()
#> ✖ tidyr::expand() masks S4Vectors::expand()
#> ✖ tidyr::extract() masks magrittr::extract()
#> ✖ metid::filter() masks metpath::filter(), dplyr::filter(), massdataset::filter(), stats::filter()
#> ✖ S4Vectors::first() masks dplyr::first()
#> ✖ xcms::groups() masks dplyr::groups()
#> ✖ dplyr::lag() masks stats::lag()
#> ✖ purrr::map() masks mixOmics::map()
#> ✖ BiocGenerics::Position() masks ggplot2::Position(), base::Position()
#> ✖ purrr::reduce() masks MSnbase::reduce()
#> ✖ S4Vectors::rename() masks dplyr::rename(), massdataset::rename()
#> ✖ lubridate::second() masks S4Vectors::second()
#> ✖ lubridate::second<-() masks S4Vectors::second<-()
#> ✖ MASS::select() masks dplyr::select(), massdataset::select()
#> ✖ purrr::set_names() masks magrittr::set_names()
#> ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
load("statistical_analysis/object_final")
object_final
#> --------------------
#> massdataset version: 1.0.34
#> --------------------
#> 1.expression_data:[ 208 x 259 data.frame]
#> 2.sample_info:[ 259 x 11 data.frame]
#> 259 samples:sample_06 sample_103 sample_11 ... sample_QC_38 sample_QC_39
#> 3.variable_info:[ 208 x 12 data.frame]
#> 208 variables:M86T95_POS M95T100_1_POS M100T160_POS ... M478T658_NEG M480T641_NEG
#> 4.sample_info_note:[ 11 x 2 data.frame]
#> 5.variable_info_note:[ 12 x 2 data.frame]
#> 6.ms2_data:[ 2134 variables x 1939 MS2 spectra]
#> --------------------
#> Processing information
#> 24 processings in total
#> Latest 3 processings show
#> slice_head ----------
#> Package Function.used Time
#> 1 massdataset slice_head() 2024-09-25 21:10:40.992678
#> 2 massdataset slice_head() 2024-09-25 21:10:41.167041
#> mutate_fc ----------
#> Package Function.used Time
#> 1 massdataset mutate_fc() 2024-09-25 21:10:41
#> mutate_p_value ----------
#> Package Function.used Time
#> 1 massdataset mutate_p_value() 2024-09-25 21:10:41
Pathway enrichment
All the results will be placed in a folder named as pathway_enrichment.
dir.create(path = "pathway_enrichment", showWarnings = FALSE)
diff_metabolites <-
object_final %>%
activate_mass_dataset(what = "variable_info") %>%
filter(p_value_adjust < 0.05) %>%
extract_variable_info()
head(diff_metabolites)
#> variable_id mz rt na_freq na_freq.1 na_freq.2 na_freq_2
#> 1 M86T95_POS 86.09716 94.57264 0.02564103 0.55454545 0.29090909 NA
#> 2 M95T100_1_POS 95.04975 99.77637 0.00000000 0.00000000 0.00000000 NA
#> 3 M104T51_POS 104.10746 51.27993 0.00000000 0.07272727 0.00000000 NA
#> 4 M113T81_POS 113.03501 80.73506 0.00000000 0.00000000 0.00000000 NA
#> 5 M113T187_POS 113.06018 186.56470 0.02564103 0.01818182 0.02727273 NA
#> 6 M114T52_POS 114.06658 52.31072 0.00000000 0.00000000 0.00000000 NA
#> na_freq.1_2 na_freq.2_2 fc p_value p_value_adjust
#> 1 NA NA 1.714629 3.034938e-20 4.912294e-19
#> 2 NA NA 1.439791 6.761432e-31 3.515945e-29
#> 3 NA NA 1.751085 2.695171e-08 1.334751e-07
#> 4 NA NA 1.500756 5.966911e-22 1.241118e-20
#> 5 NA NA 1.951417 1.848299e-17 2.023401e-16
#> 6 NA NA 1.382309 9.673602e-05 3.095553e-04
#> Compound.name CAS.ID HMDB.ID KEGG.ID Lab.ID Adduct mz.error
#> 1 Piperidine 110-89-4 <NA> C01746 MONA_2852 (M+H)+ 1.7468691
#> 2 Phenol 108-95-2 <NA> D01960 MONA_18506 (M+H)+ 1.4164283
#> 3 5-Amino-1-pentanol 2508-29-4 <NA> <NA> NO07238 (M+H)+ 1.1691284
#> 4 Uracil 66-22-8 HMDB00300 C00106 NO07292 (M+H)+ 1.2189636
#> 5 1,4-Cyclohexanedione <NA> <NA> <NA> MONA_14519 (M+H)+ 1.0516262
#> 6 Creatinine 60-27-5 HMDB00562 C00791 NO07303 (M+H)+ 0.7404486
#> mz.match.score RT.error RT.match.score CE SS
#> 1 0.9932417 NA NA 30 0.6143541
#> 2 0.9955515 NA NA 10 0.6102452
#> 3 0.9969671 NA NA 5 0.5971697
#> 4 0.9967035 NA NA 10 0.6484578
#> 5 0.9975454 NA NA HCD (NCE 20-30-40%) 0.5401414
#> 6 0.9987824 NA NA 20 0.6094410
#> Total.score Database Level
#> 1 0.6996930 MoNA_0.0.1 2
#> 2 0.6979976 MoNA_0.0.1 2
#> 3 0.6906242 NIST_0.0.1 2
#> 4 0.7213092 NIST_0.0.1 2
#> 5 0.6566000 MoNA_0.0.1 2
#> 6 0.6985921 NIST_0.0.1 2
Load KEGG human pathway database
data("kegg_hsa_pathway", package = "metpath")
kegg_hsa_pathway
#> ---------Pathway source&version---------
#> KEGG & 2021-12-13
#> -----------Pathway information------------
#> 345 pathways
#> 334 pathways have genes
#> 0 pathways have proteins
#> 281 pathways have compounds
#> Pathway class (top 10): Metabolism; Carbohydrate metabolism;Metabolism; Lipid metabolism
get_pathway_class(kegg_hsa_pathway)
#> # A tibble: 43 × 2
#> class n
#> <chr> <int>
#> 1 Cellular Processes; Cell growth and death 8
#> 2 Cellular Processes; Cell motility 1
#> 3 Cellular Processes; Cellular community - eukaryotes 5
#> 4 Cellular Processes; Transport and catabolism 7
#> 5 Environmental Information Processing; Membrane transport 1
#> 6 Environmental Information Processing; Signal transduction 26
#> 7 Environmental Information Processing; Signaling molecules and interact… 5
#> 8 Genetic Information Processing; Folding, sorting and degradation 7
#> 9 Genetic Information Processing; Replication and repair 7
#> 10 Genetic Information Processing; Transcription 3
#> # ℹ 33 more rows
Remove the disease pathways:
#get the class of pathways
pathway_class =
metpath::pathway_class(kegg_hsa_pathway)
head(pathway_class)
#> $hsa00010
#> [1] "Metabolism; Carbohydrate metabolism"
#>
#> $hsa00020
#> [1] "Metabolism; Carbohydrate metabolism"
#>
#> $hsa00030
#> [1] "Metabolism; Carbohydrate metabolism"
#>
#> $hsa00040
#> [1] "Metabolism; Carbohydrate metabolism"
#>
#> $hsa00051
#> [1] "Metabolism; Carbohydrate metabolism"
#>
#> $hsa00052
#> [1] "Metabolism; Carbohydrate metabolism"
remain_idx =
pathway_class %>%
unlist() %>%
stringr::str_detect("Disease") %>%
`!`() %>%
which()
remain_idx
#> [1] 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
#> [19] 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
#> [37] 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54
#> [55] 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72
#> [73] 73 74 75 76 77 78 79 80 81 82 83 84 85 90 91 92 93 94
#> [91] 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112
#> [109] 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130
#> [127] 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148
#> [145] 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166
#> [163] 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184
#> [181] 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202
#> [199] 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220
#> [217] 221 222 223 224 225 226 227 228 229 230 236 240 241 242 243 244 245 246
#> [235] 247 248 249 250 251 252 253 254
pathway_database =
kegg_hsa_pathway[remain_idx]
pathway_database
#> ---------Pathway source&version---------
#> KEGG & 2021-12-13
#> -----------Pathway information------------
#> 242 pathways
#> 235 pathways have genes
#> 0 pathways have proteins
#> 191 pathways have compounds
#> Pathway class (top 10): Metabolism; Carbohydrate metabolism;Metabolism; Lipid metabolism
kegg_id <-
diff_metabolites$KEGG.ID
kegg_id <-
kegg_id[!is.na(kegg_id)]
kegg_id
#> [1] "C01746" "D01960" "C00106" "C00791" "C00906" "C00262" "C14790" "C02237"
#> [9] "C00073" "C00637" "C16366" "C07481" "C17846" "C14214" "C01595" "C00410"
#> [17] "C00762" "C00735" "C17337" "C01921" "C04230" "C04230" "C06539" "C00186"
#> [25] "C01546" "C01732" "C02226" "C00064" "C06104" "C07599" "0" "C01601"
#> [33] "C00366" "C08322" "C16308" "C00294" "C03343" "C10911" "C05498" "C05472"
#> [41] "C04555"
result <-
enrich_kegg(query_id = kegg_id,
query_type = "compound",
id_type = "KEGG",
pathway_database = pathway_database,
p_cutoff = 0.05,
p_adjust_method = "BH",
threads = 3)
#> 191 pathways.
Check the result:
result
#> ---------Pathway database&version---------
#> KEGG & 1.0.8
#> -----------Enrichment result------------
#> 191 pathways are enriched
#> 8 pathways p-values < 0.05
#> Steroid hormone biosynthesis
#> Aldosterone-regulated sodium reabsorption
#> Cushing syndrome
#> Purine metabolism
#> Caffeine metabolism ... (only top 5 shows)
#> -----------Parameters------------
#> Package Function.used Time
#> 1 metpath enrich_kegg() 2024-09-25 21:11:19
Plot to show pathway enrichment result
enrich_bar_plot(object = result,
x_axis = "p_value",
cutoff = 0.05)
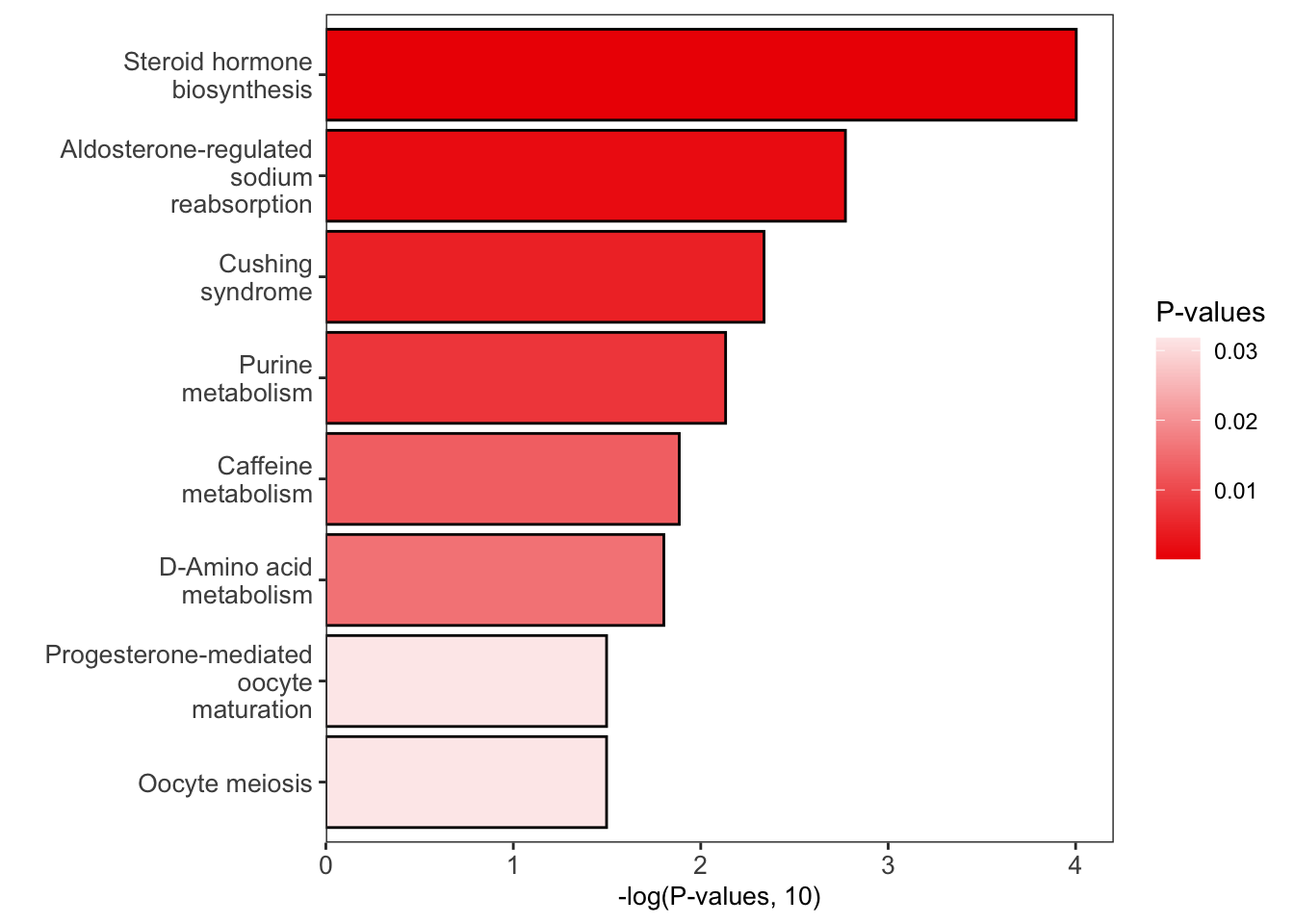
enrich_scatter_plot(object = result, y_axis = "p_value", y_axis_cutoff = 0.05)
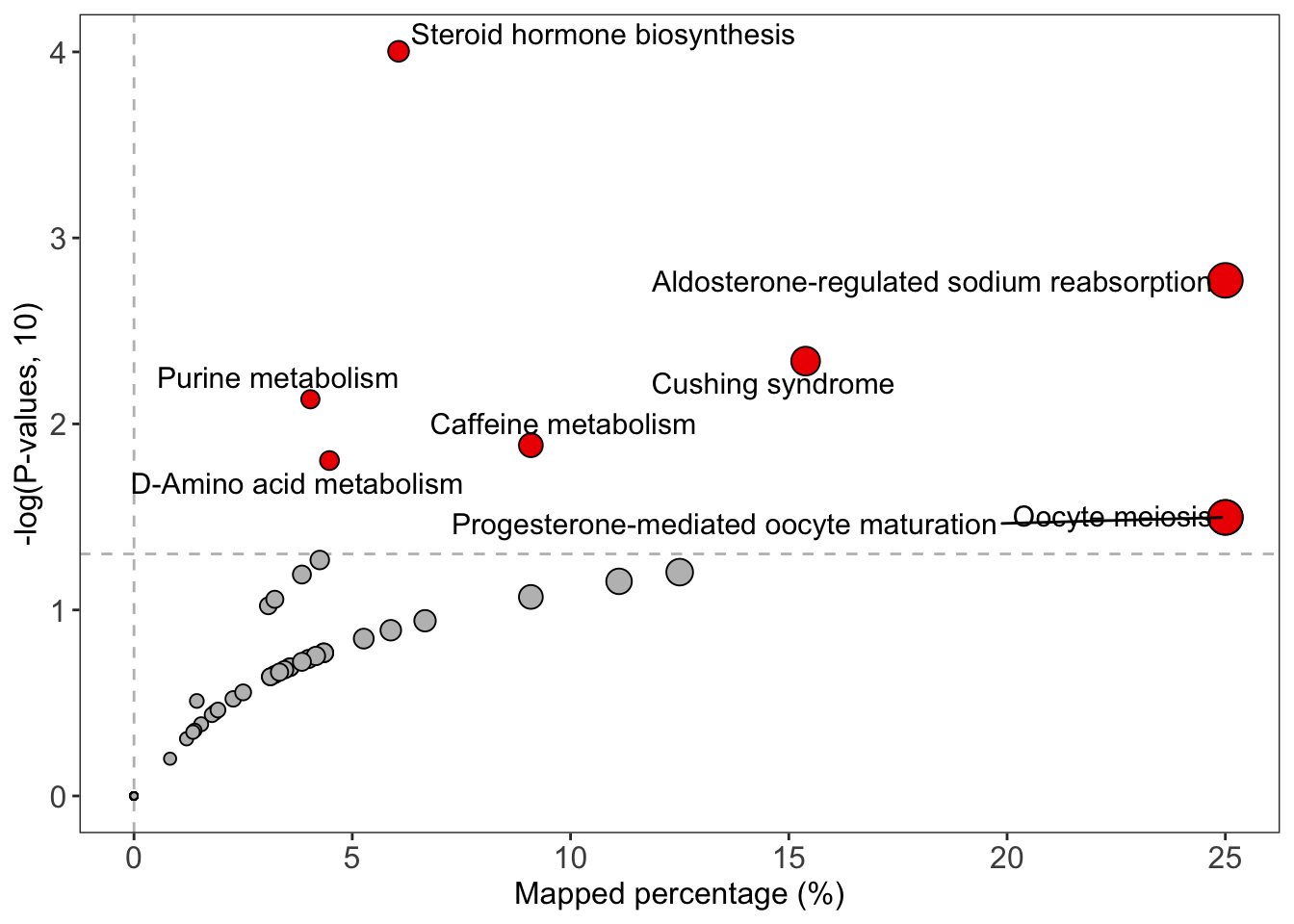
enrich_network(
object = result,
point_size = "p_value",
p_cutoff = 0.05,
only_significant_pathway = TRUE
)
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.0
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1
#> [4] purrr_1.0.2 readr_2.1.5 tibble_3.2.1
#> [7] tidyverse_2.0.0 metid_1.2.34 metpath_1.0.8
#> [10] ComplexHeatmap_2.20.0 mixOmics_6.28.0 lattice_0.22-6
#> [13] MASS_7.3-61 massstat_1.0.6 tidyr_1.3.1
#> [16] ggfortify_0.4.17 massqc_1.0.7 masscleaner_1.0.12
#> [19] MSnbase_2.30.1 ProtGenerics_1.36.0 S4Vectors_0.42.1
#> [22] Biobase_2.64.0 BiocGenerics_0.50.0 mzR_2.38.0
#> [25] Rcpp_1.0.13 xcms_4.2.3 BiocParallel_1.38.0
#> [28] massprocesser_1.0.10 ggplot2_3.5.1 dplyr_1.1.4
#> [31] magrittr_2.0.3 masstools_1.0.13 massdataset_1.0.34
#> [34] tidymass_1.0.9
#>
#> loaded via a namespace (and not attached):
#> [1] fs_1.6.4 matrixStats_1.3.0
#> [3] bitops_1.0-8 fit.models_0.64
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 tools_4.4.1
#> [9] doRNG_1.8.6 backports_1.5.0
#> [11] utf8_1.2.4 R6_2.5.1
#> [13] lazyeval_0.2.2 GetoptLong_1.0.5
#> [15] withr_3.0.1 prettyunits_1.2.0
#> [17] gridExtra_2.3 preprocessCore_1.66.0
#> [19] cli_3.6.3 fastDummies_1.7.4
#> [21] labeling_0.4.3 sass_0.4.9
#> [23] mvtnorm_1.3-1 robustbase_0.99-4
#> [25] randomForest_4.7-1.1 proxy_0.4-27
#> [27] pbapply_1.7-2 foreign_0.8-87
#> [29] rrcov_1.7-6 MetaboCoreUtils_1.12.0
#> [31] parallelly_1.38.0 itertools_0.1-3
#> [33] limma_3.60.4 readxl_1.4.3
#> [35] rstudioapi_0.16.0 impute_1.78.0
#> [37] generics_0.1.3 shape_1.4.6.1
#> [39] zip_2.3.1 Matrix_1.7-0
#> [41] MALDIquant_1.22.3 fansi_1.0.6
#> [43] abind_1.4-5 lifecycle_1.0.4
#> [45] yaml_2.3.10 SummarizedExperiment_1.34.0
#> [47] SparseArray_1.4.8 crayon_1.5.3
#> [49] PSMatch_1.8.0 KEGGREST_1.44.1
#> [51] pillar_1.9.0 knitr_1.48
#> [53] GenomicRanges_1.56.1 rjson_0.2.22
#> [55] corpcor_1.6.10 codetools_0.2-20
#> [57] glue_1.7.0 pcaMethods_1.96.0
#> [59] data.table_1.16.0 remotes_2.5.0
#> [61] MultiAssayExperiment_1.30.3 vctrs_0.6.5
#> [63] png_0.1-8 cellranger_1.1.0
#> [65] gtable_0.3.5 cachem_1.1.0
#> [67] xfun_0.47 openxlsx_4.2.7
#> [69] S4Arrays_1.4.1 tidygraph_1.3.1
#> [71] pcaPP_2.0-5 ncdf4_1.23
#> [73] iterators_1.0.14 statmod_1.5.0
#> [75] robust_0.7-5 progress_1.2.3
#> [77] GenomeInfoDb_1.40.1 rprojroot_2.0.4
#> [79] bslib_0.8.0 affyio_1.74.0
#> [81] rpart_4.1.23 colorspace_2.1-1
#> [83] DBI_1.2.3 Hmisc_5.1-3
#> [85] nnet_7.3-19 tidyselect_1.2.1
#> [87] compiler_4.4.1 MassSpecWavelet_1.70.0
#> [89] htmlTable_2.4.3 DelayedArray_0.30.1
#> [91] plotly_4.10.4 bookdown_0.40
#> [93] checkmate_2.3.2 scales_1.3.0
#> [95] DEoptimR_1.1-3 affy_1.82.0
#> [97] digest_0.6.37 rmarkdown_2.28
#> [99] XVector_0.44.0 htmltools_0.5.8.1
#> [101] pkgconfig_2.0.3 base64enc_0.1-3
#> [103] MatrixGenerics_1.16.0 highr_0.11
#> [105] fastmap_1.2.0 rlang_1.1.4
#> [107] GlobalOptions_0.1.2 htmlwidgets_1.6.4
#> [109] UCSC.utils_1.0.0 farver_2.1.2
#> [111] jquerylib_0.1.4 jsonlite_1.8.8
#> [113] MsExperiment_1.6.0 mzID_1.42.0
#> [115] RCurl_1.98-1.16 Formula_1.2-5
#> [117] GenomeInfoDbData_1.2.12 patchwork_1.2.0
#> [119] munsell_0.5.1 viridis_0.6.5
#> [121] MsCoreUtils_1.16.1 vsn_3.72.0
#> [123] furrr_0.3.1 stringi_1.8.4
#> [125] ggraph_2.2.1 zlibbioc_1.50.0
#> [127] plyr_1.8.9 parallel_4.4.1
#> [129] listenv_0.9.1 ggrepel_0.9.5
#> [131] Biostrings_2.72.1 MsFeatures_1.12.0
#> [133] graphlayouts_1.1.1 hms_1.1.3
#> [135] Spectra_1.14.1 circlize_0.4.16
#> [137] igraph_2.0.3 QFeatures_1.14.2
#> [139] rngtools_1.5.2 reshape2_1.4.4
#> [141] XML_3.99-0.17 evaluate_0.24.0
#> [143] blogdown_1.19 BiocManager_1.30.25
#> [145] tzdb_0.4.0 foreach_1.5.2
#> [147] missForest_1.5 tweenr_2.0.3
#> [149] polyclip_1.10-7 future_1.34.0
#> [151] clue_0.3-65 ggforce_0.4.2
#> [153] AnnotationFilter_1.28.0 e1071_1.7-14
#> [155] RSpectra_0.16-2 ggcorrplot_0.1.4.1
#> [157] viridisLite_0.4.2 class_7.3-22
#> [159] rARPACK_0.11-0 memoise_2.0.1
#> [161] ellipse_0.5.0 IRanges_2.38.1
#> [163] cluster_2.1.6 timechange_0.3.0
#> [165] globals_0.16.3 here_1.0.1