介绍
如果你想为 metID 构建数据库,可以使用 massDatabase 包。massdatabase 是一个 R 包,用于操作在线公共数据库,并结合其他工具进行化合物注释和通路富集分析。massdatabase 是一个灵活、简单且强大的工具,可以在所有平台上安装,使用户能够利用所有在线公共数据库进行生物功能挖掘。
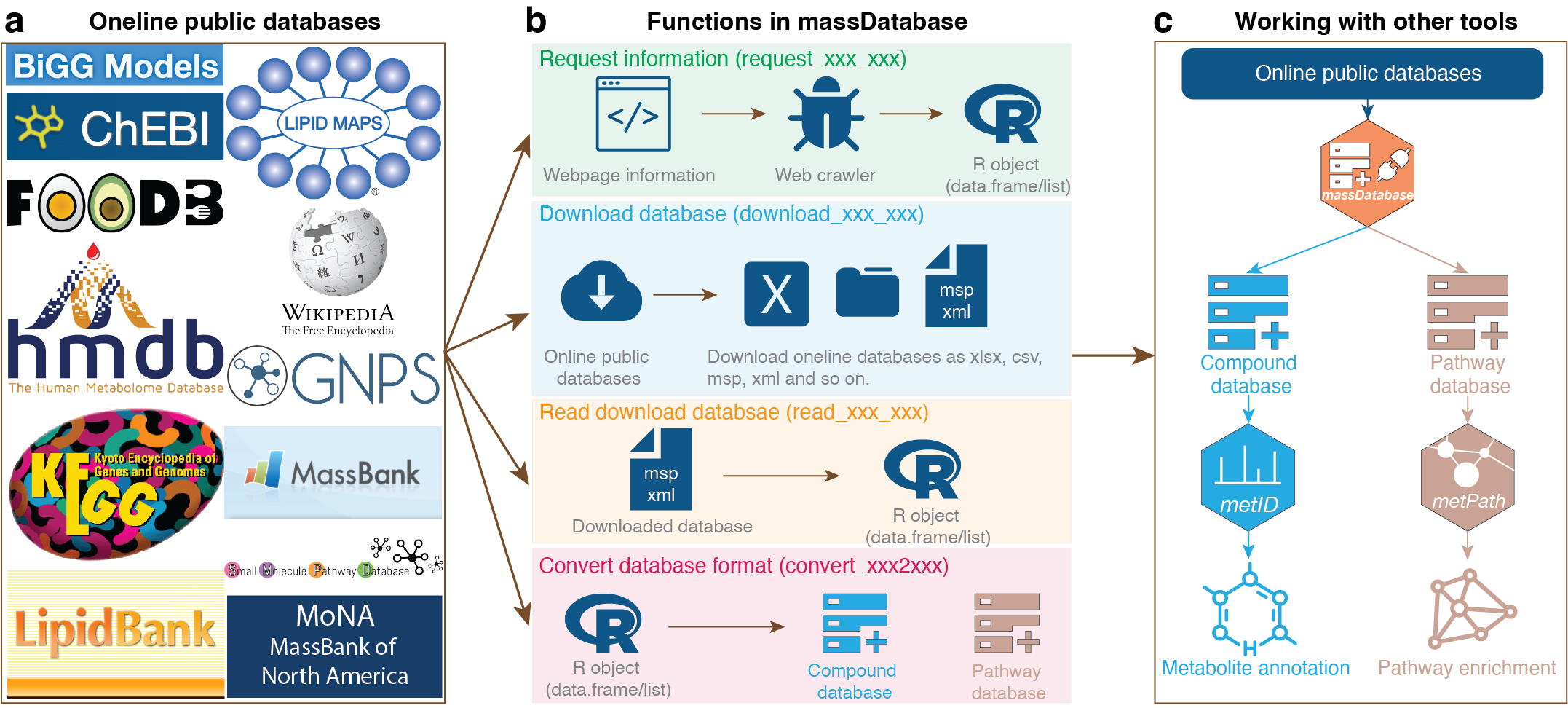
安装 massdatabase
你可以从 GitLab 安装 massdatabase。
if(!require(remotes)){
install.packages("remotes")
}
remotes::install_gitlab("tidymass/massdatabase")
或从 GitHub 安装
remotes::install_github("tidymass/massdatabase")
或者从 tidymass.org 安装
source("https://www.tidymass.org/tidymass-packages/install_tidymass.txt")
install_tidymass(from = "tidymass.org", which_package = "massdatabase")
BIGG 数据库
BIGG model 是一个基于基因组规模的代谢网络重建知识库。
下载 BIGG 通用代谢物数据库:
library(massdatabase)
#> massdatabase 1.0.10 (2024-09-25 19:56:56.178093)
#>
#> Attaching package: 'massdatabase'
#> The following object is masked _by_ 'package:masstools':
#>
#> show_progresser
download_bigg_universal_metabolite(path = "database_construction/",
sleep = 1)
然后读取并将其转换为 databaseClass 格式。
data <-
read_bigg_universal_metabolite(path = ".")
下载数据库可能需要一段时间。
然后将其转换为 databaseClass 格式。
bigg_database <-
convert_bigg_universal2metid(data = data, path = ".")
bigg_database
你可以将数据库保存以供以后使用。
save(bigg_database, file = "database_construction/bigg_database")
ChEBI 数据库
Chemical Entities of Biological Interest (ChEBI) 是一个专注于“小”化学化合物的分子实体的免费字典。
下载 ChEBI 化合物数据库:
library(massdatabase)
download_chebi_compound(path = "database_construction/")
然后读取并将其转换为 databaseClass 格式。
data <-
read_chebi_compound(path = "database_construction")
#> Reading data...
然后将其转换为 databaseClass 格式。
chebi_database <-
convert_chebi2metid(data = data, path = "database_construction")
#> No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c044148807
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c044148807
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
chebi_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:CHEBI
#> Link:https://www.ebi.ac.uk/chebi/init.do
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 34 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 152379 metabolites in total.
#> 0 metabolites with spectra in positive mode.
#> 0 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:0
#>
#> Collision energy in negative mode:
#> Total number:0
#>
你可以将数据库保存以供以后使用。
save(chebi_database, file = "database_construction/chebi_database")
FooDB 数据库
FooDB 是全球最大且最全面的食物成分、化学和生物学资源。
下载 FooDB 数据库:
library(massdatabase)
download_foodb_compound(compound_id = "all", path = "database_construction/")
下载数据库可能需要一段时间。
然后读取并将其转换为 databaseClass 格式。
data <-
read_foodb_compound(path = "database_construction")
然后将其转换为 databaseClass 格式。
foodb_dataabse <-
convert_foodb2metid(data = data, path = "database_construction")
foodb_dataabse
你可以将数据库保存以供以后使用。
save(foodb_dataabse, file = "database_construction/foodb_dataabse")
GNPS 数据库
GNPS 是一个基于网络的质谱生态系统,旨在成为全社区范围内的开放访问知识库,用于组织和共享原始、处理过的或带有注释的质谱数据(MS/MS)。GNPS 在数据的整个生命周期内(从最初的数据采集/分析到发布后)都有助于鉴定和发现。
下载数据库:
library(massdatabase)
download_gnps_spectral_library(gnps_library = "HMDB",
path = "database_construction")
参数 gnps_library 应是此网站上的 GNPS Library 之一。
https://gnps-external.ucsd.edu/gnpslibrary
下载数据库可能需要一段时间。
然后读取并将其转换为 databaseClass 格式。
data <-
read_msp_data_gnps(file = "database_construction/HMDB.msp")
#>
indexing HMDB.msp [=======================================] 274.90GB/s, eta: 0s
#> 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
然后将其转换为 databaseClass 格式。
gnps_database <-
convert_gnps2metid(data = data, path = "database_construction/")
#> Extracting MS1 inforamtion...
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Done.
#> Extracting MS2 inforamtion...
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Done.
#> Organizing...
#> Calculating accurate mass...
#> Done.
#> 1% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c035e86342
#>
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c035e86342
#>
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
gnps_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:GNPS
#> Link:https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 25 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 2235 metabolites in total.
#> 1907 metabolites with spectra in positive mode.
#> 328 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:1
#> Unknown_1
#> Collision energy in negative mode:
#> Total number:1
#> Unknown_1
你可以将数据库保存以供以后使用。
save(gnps_database, file = "database_construction/gnps_database")
KEGG 数据库
KEGG 是一个数据库资源,用于从分子层面信息中理解生物系统(如细胞、个体和生态系统)的高级功能和用途,特别是通过基因组测序和其他高通量实验技术生成的大规模分子数据集。
下载数据库:
library(massdatabase)
download_kegg_compound(path = "database_construction/")
下载数据库可能需要一段时间。
然后读取并将其转换为 `database
Class` 格式。
data <-
read_kegg_compound(path = "database_construction/")
然后将其转换为 databaseClass 格式。
kegg_database <-
convert_kegg2metid(data = data, path = "database_construction")
kegg_database
你可以将数据库保存以供以后使用。
save(kegg_database, file = "database_construction/kegg_database")
LipidMaps 数据库
LIPID MAPS Lipidomics Gateway 创建于2003年,通过 NIH 的“胶水资助”项目,为国际脂质研究社区提供脂质命名法、数据库、工具、标准、教程等资源。
下载数据库:
library(massdatabase)
download_lipidmaps_lipid(path = "database_construction")
下载数据库可能需要一段时间。
解压下载的 zip 文件。
unzip(zipfile = "database_construction/LMSD.sdf.zip",
exdir = "database_construction/")
然后读取并将其转换为 databaseClass 格式。
data <-
read_sdf_data_lipidmaps(file = "database_construction/structures.sdf")
#> Reading data, it may take a while...
#> Done.
#> Organizing...
#> Done.
然后将其转换为 databaseClass 格式。
lipidmaps_database <-
convert_lipidmaps2metid(data = data, path = "database_construction/")
#> Organizing...
#> No POS file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c04dc6320c
#> No NEG file in your /var/folders/hh/628g4x5n0ygcv7ql3lsbl1z80000gn/T//Rtmpa1hD39/file55c04dc6320c
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading negative MS2 data...
#> Matching metabolites with MS2 spectra (positive)...
#> Matching metabolites with MS2 spectra (negative)...
#> All done!
lipidmaps_database
#> -----------Base information------------
#> Version:2024-09-25
#> Source:LIPIDMAPS
#> Link:https://www.lipidmaps.org/
#> Creater:Xiaotao Shen(shenxt@stanford.edu)
#> Without RT informtaion
#> -----------Spectral information------------
#> 27 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 47500 metabolites in total.
#> 0 metabolites with spectra in positive mode.
#> 0 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:0
#>
#> Collision energy in negative mode:
#> Total number:0
#>
你可以将数据库保存以供以后使用。
save(lipidmaps_database, file = "database_construction/lipidmaps_database")
MassBank 数据库
MassBank 是一个社区合作项目,欢迎大家贡献。请参考我们的贡献文档,并通过 GitHub 或电子邮件与我们联系。
下载数据库:
library(massdatabase)
download_massbank_compound(source = "nist", path = "database_construction/")
下载数据库可能需要一段时间。
然后读取并将其转换为 databaseClass 格式。
data <- read_msp_data_massbank(file = "database_construction/massbank_compound/MassBank_NIST.msp")
#> 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
读取数据可能需要一段时间。
然后将其转换为 databaseClass 格式。
massbank_database <-
convert_massbank2metid_nist(data = data, path = "database_construction/")
转换数据可能需要一段时间。
massbank_database
你可以将数据库保存以供以后使用。
save(massbank_database, file = "database_construction/massbank_database")
SMPDB 数据库
SMPDB 是一个交互式、可视化的数据库,包含30000多个仅在人体中发现的小分子通路。
下载数据库:
library(massdatabase)
download_smpdb_pathway(path = "database_construction/")
下载数据库可能需要一段时间。
然后读取并将其转换为 databaseClass 格式。
data <-
read_smpdb_pathway(path = "database_construction/", only_primarity_pathway = TRUE)
然后将其转换为 databaseClass 格式。
smpdb_pathway_database <-
convert_smpdb2metpath(data = data, path = "database_construction/")
smpdb_pathway_database
你可以将数据库保存以供以后使用。
save(smpdb_pathway_database, file = "database_construction/smpdb_pathway_database")
引用
如果你在研究中使用了 massdatabase,请引用以下论文:
会话信息
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.0
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] MSnbase_2.30.1 ProtGenerics_1.36.0 S4Vectors_0.42.1
#> [4] mzR_2.38.0 Rcpp_1.0.13 Biobase_2.64.0
#> [7] BiocGenerics_0.50.0 massdataset_1.0.34 metid_1.2.34
#> [10] metpath_1.0.8 ggplot2_3.5.1 dplyr_1.1.4
#> [13] magrittr_2.0.3 masstools_1.0.13 massdatabase_1.0.10
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-8 tibble_3.2.1
#> [3] cellranger_1.1.0 polyclip_1.10-7
#> [5] preprocessCore_1.66.0 XML_3.99-0.17
#> [7] lifecycle_1.0.4 doParallel_1.0.17
#> [9] rprojroot_2.0.4 globals_0.16.3
#> [11] lattice_0.22-6 vroom_1.6.5
#> [13] MASS_7.3-61 MultiAssayExperiment_1.30.3
#> [15] openxlsx_4.2.7 limma_3.60.4
#> [17] plotly_4.10.4 sass_0.4.9
#> [19] rmarkdown_2.28 jquerylib_0.1.4
#> [21] yaml_2.3.10 remotes_2.5.0
#> [23] zip_2.3.1 MsCoreUtils_1.16.1
#> [25] pbapply_1.7-2 DBI_1.2.3
#> [27] RColorBrewer_1.1-3 abind_1.4-5
#> [29] zlibbioc_1.50.0 rvest_1.0.4
#> [31] GenomicRanges_1.56.1 purrr_1.0.2
#> [33] AnnotationFilter_1.28.0 ggraph_2.2.1
#> [35] RCurl_1.98-1.16 tweenr_2.0.3
#> [37] circlize_0.4.16 GenomeInfoDbData_1.2.12
#> [39] IRanges_2.38.1 ggrepel_0.9.5
#> [41] listenv_0.9.1 parallelly_1.38.0
#> [43] ncdf4_1.23 codetools_0.2-20
#> [45] DelayedArray_0.30.1 DT_0.33
#> [47] xml2_1.3.6 ggforce_0.4.2
#> [49] tidyselect_1.2.1 shape_1.4.6.1
#> [51] UCSC.utils_1.0.0 farver_2.1.2
#> [53] viridis_0.6.5 base64enc_0.1-3
#> [55] matrixStats_1.3.0 jsonlite_1.8.8
#> [57] GetoptLong_1.0.5 tidygraph_1.3.1
#> [59] iterators_1.0.14 foreach_1.5.2
#> [61] tools_4.4.1 progress_1.2.3
#> [63] stringdist_0.9.12 glue_1.7.0
#> [65] gridExtra_2.3 SparseArray_1.4.8
#> [67] xfun_0.47 here_1.0.1
#> [69] MatrixGenerics_1.16.0 GenomeInfoDb_1.40.1
#> [71] withr_3.0.1 BiocManager_1.30.25
#> [73] fastmap_1.2.0 fansi_1.0.6
#> [75] blogdown_1.19 digest_0.6.37
#> [77] R6_2.5.1 colorspace_2.1-1
#> [79] rsvg_2.6.1 utf8_1.2.4
#> [81] tidyr_1.3.1 generics_0.1.3
#> [83] data.table_1.16.0 prettyunits_1.2.0
#> [85] graphlayouts_1.1.1 PSMatch_1.8.0
#> [87] httr_1.4.7 htmlwidgets_1.6.4
#> [89] S4Arrays_1.4.1 pkgconfig_2.0.3
#> [91] gtable_0.3.5 ComplexHeatmap_2.20.0
#> [93] impute_1.78.0 XVector_0.44.0
#> [95] furrr_0.3.1 htmltools_0.5.8.1
#> [97] bookdown_0.40 MALDIquant_1.22.3
#> [99] clue_0.3-65 scales_1.3.0
#> [101] png_0.1-8 knitr_1.48
#> [103] rstudioapi_0.16.0 tzdb_0.4.0
#> [105] reshape2_1.4.4 rjson_0.2.22
#> [107] curl_5.2.2 cachem_1.1.0
#> [109] GlobalOptions_0.1.2 stringr_1.5.1
#> [111] parallel_4.4.1 mzID_1.42.0
#> [113] vsn_3.72.0 pillar_1.9.0
#> [115] grid_4.4.1 vctrs_0.6.5
#> [117] pcaMethods_1.96.0 cluster_2.1.6
#> [119] evaluate_0.24.0 readr_2.1.5
#> [121] cli_3.6.3 compiler_4.4.1
#> [123] rlang_1.1.4 crayon_1.5.3
#> [125] Rdisop_1.64.0 QFeatures_1.14.2
#> [127] ChemmineR_3.56.0 affy_1.82.0
#> [129] plyr_1.8.9 stringi_1.8.4
#> [131] viridisLite_0.4.2 BiocParallel_1.38.0
#> [133] munsell_0.5.1 Biostrings_2.72.1
#> [135] lazyeval_0.2.2 Matrix_1.7-0
#> [137] hms_1.1.3 bit64_4.0.5
#> [139] future_1.34.0 KEGGREST_1.44.1
#> [141] statmod_1.5.0 SummarizedExperiment_1.34.0
#> [143] igraph_2.0.3 memoise_2.0.1
#> [145] affyio_1.74.0 bslib_0.8.0
#> [147] bit_4.0.5 readxl_1.4.3