如果你有内部标准并且已经获取了 MS2 光谱数据,那么你可以使用 metID 包来构建内部 MS2 光谱数据库。
对于用户如何运行 LC/MS 数据并没有具体的要求。由于在 metID 中的内部数据库构建是为了让用户能够自行获得内部数据库(包括代谢物的 m/z、保留时间和 MS/MS 光谱,用于一级注释 (Sumner et al., 2007)),因此用户只需使用与实验室实际样品相同的色谱柱、LC 梯度和 MS 设置运行标准样品即可。
数据准备
首先,请使用 ProteoWizard 将原始标准 MS 数据(正负模式)转换为 mzXML 格式。参数设置如下图所示:
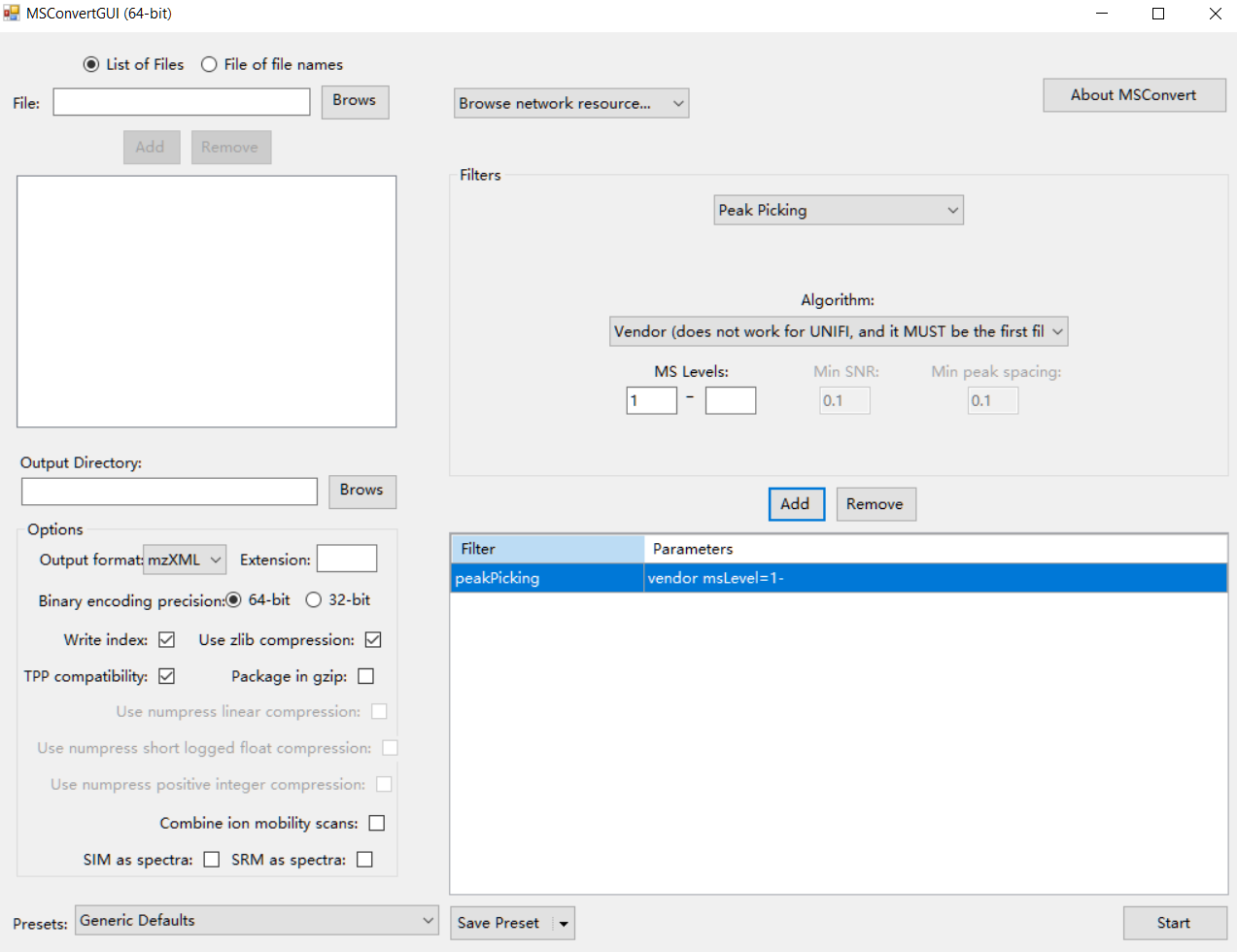
数据组织
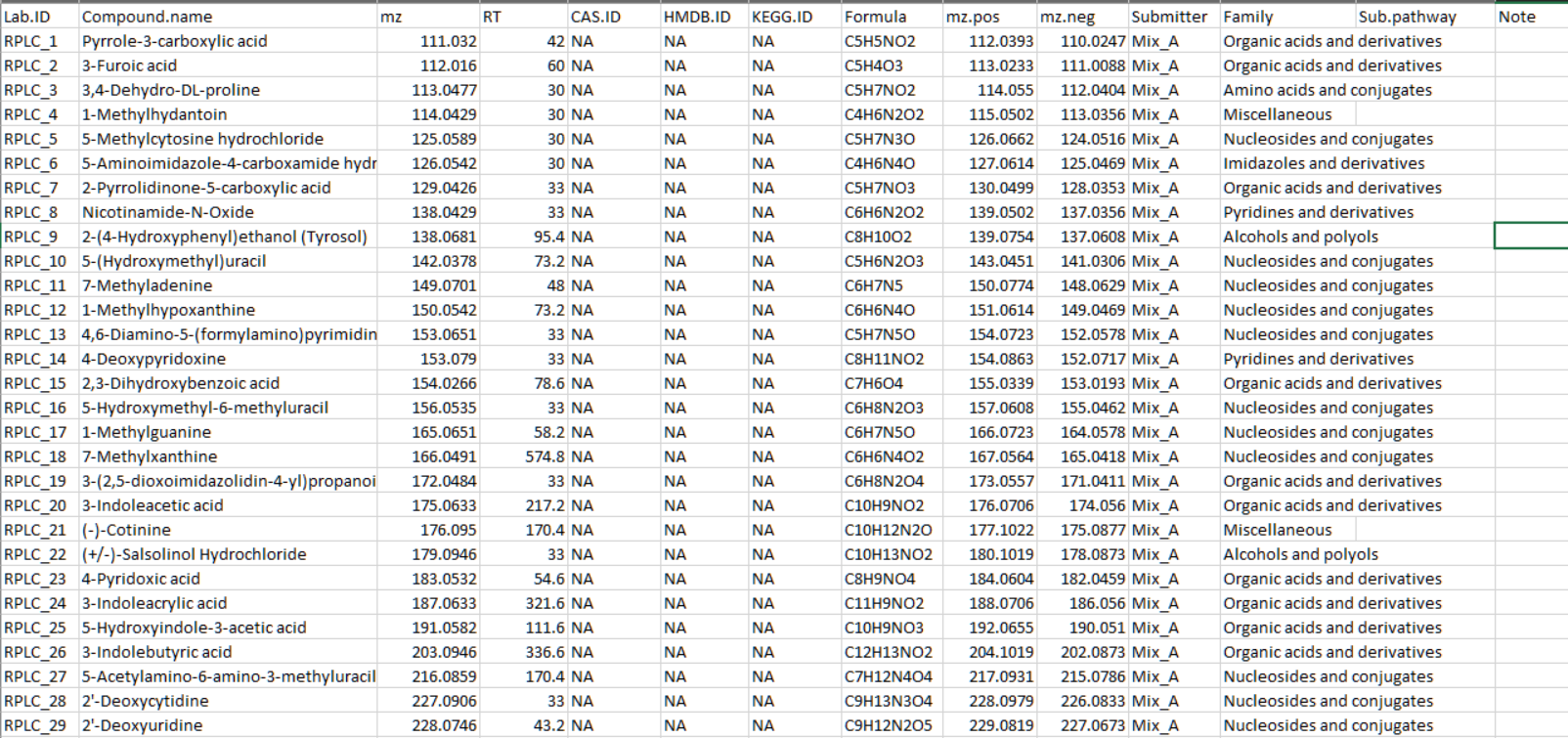
其次,请将标准信息整理成表格,并输出为 csv 或 xlsx 格式。标准信息的格式可以参考 demoData 包中的演示数据。
从第1列到第11列,依次为 “Lab.ID”、“Compound.name”、“mz”、“RT”、“CAS.ID”、“HMDB.ID”、“KEGG.ID”、“Formula”、“mz.pos”、“mz.neg”、“Submitter”。如果你有其他的标准信息也没问题。例如演示数据中还有其他附加信息:“Family”、“Sub.pathway” 和 “Note”。
-
Lab.ID: 无重复。
-
mz: 化合物的精确质量。
-
RT: 保留时间,单位为秒。
-
mz.pos: 化合物在正模式下的质量电荷比,例如 M+H。你可以将其设置为 NA。
-
mz.neg: 化合物在负模式下的质量电荷比,例如 M-H。你可以将其设置为 NA。
-
Submitter: 提交者的名字或组织名称。你可以将其设置为 NA。
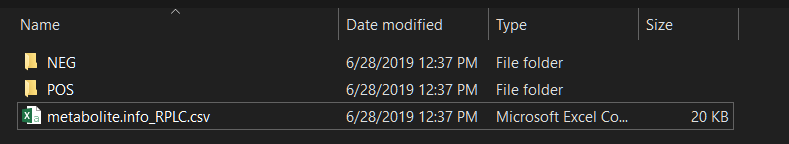
然后创建一个文件夹,将 mzXML 格式的数据集(正模式放入 ‘POS’ 文件夹,负模式放入 ‘NEG’ 文件夹)和化合物信息放入其中。mzXML 文件名应包含碰撞能量。例如 test_NCE25.mzXML。
mzXML 文件的命名应为:xxx_NCE25.mzXML。
运行 construct_database() 函数
这里我们使用 demoData 包中的演示数据来展示如何使用 construct_database() 函数来构建数据库。
我们首先准备数据集。
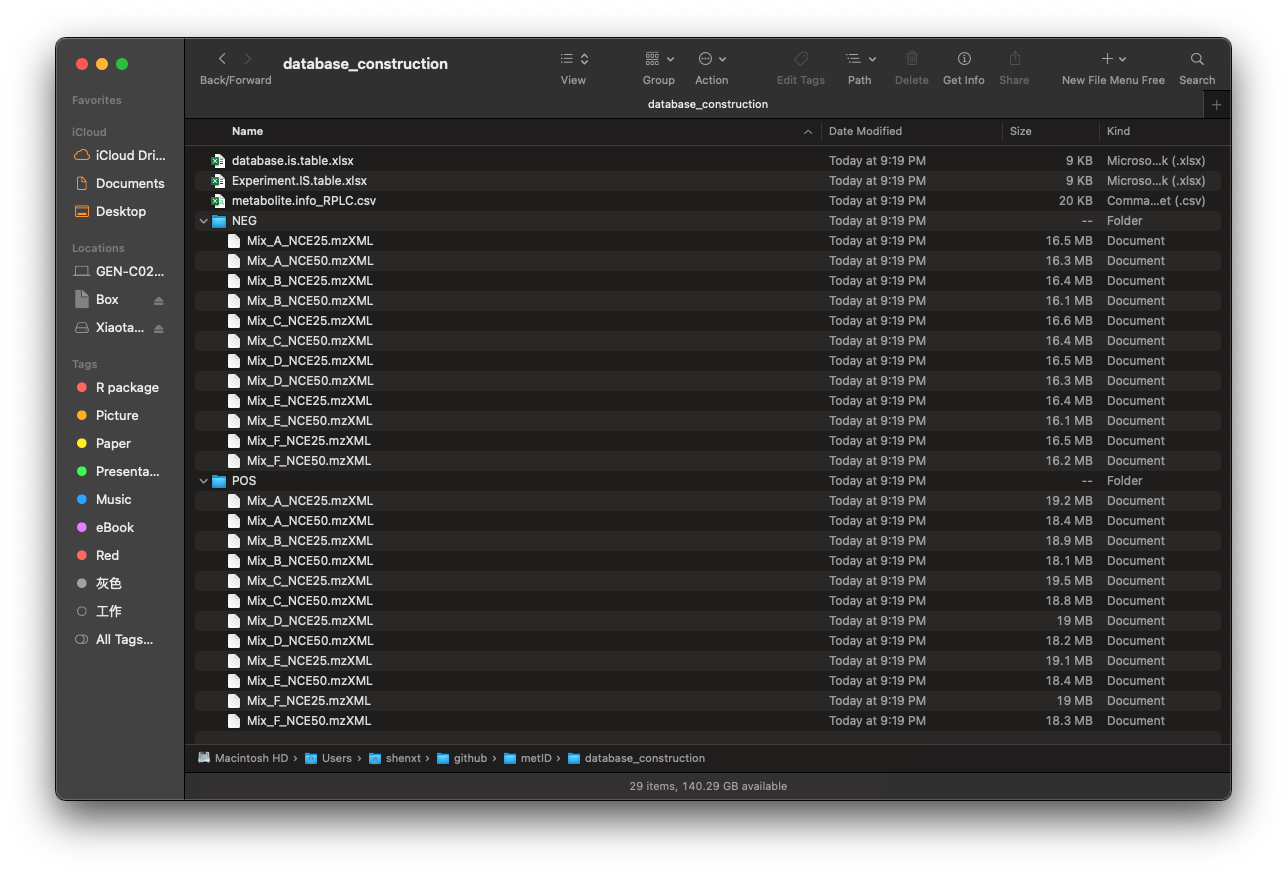
在此下载数据,然后将所有文件放入 “database_construction” 文件夹。
然后在你的工作目录中会有一个名为 database_construction 的文件夹,如下图所示:
接下来运行 construct_database() 函数,我们就可以生成数据库。
library(metid)
new.path <- file.path("./database_construction")
test.database <- construct_database(
path = new.path,
version = "0.0.1",
metabolite.info.name = "metabolite.info_RPLC.csv",
source = "Michael Snyder lab",
link = "http://snyderlab.stanford.edu/",
creater = "Xiaotao Shen",
email = "shenxt1990@163.com",
rt = TRUE,
mz.tol = 15,
rt.tol = 30,
threads = 3
)
construct_database() 函数的参数可以通过 ?construct_database() 查找。
test.database 是一个 databaseClass 对象,你可以打印它来查看其信息。
test.database
#> -----------Base information------------
#> Version:0.0.1
#> Source:Michael Snyder lab
#> Link:http://snyderlab.stanford.edu/
#> Creater:Xiaotao Shen(shenxt1990@163.com)
#> With RT information
#> -----------Spectral information------------
#> 14 items of metabolite information:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg (top10)
#> 172 metabolites in total.
#> 41 metabolites with spectra in positive mode.
#> 45 metabolites with spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number:1
#> NCE25
#> Collision energy in negative mode:
#> Total number:1
#> NCE25
注意:
test.database只是一个演示数据库(metIdentifyClass 对象)。我们不会将其用于后续的代谢物鉴定。请将此数据库保存在你的本地文件夹中,并确保保存的文件名和数据库名称一致。例如:
save(test.database, file = "test.database")
如果你使用不同的名称保存
test.database,在使用时会出现错误。
MS1 数据库
如果你没有 MS2 数据,你仍然可以使用 construct_database() 函数来构建 MS1 数据库。
会话信息
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS Sonoma 14.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Asia/Singapore
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] MSnbase_2.30.1 ProtGenerics_1.36.0 S4Vectors_0.42.1
#> [4] mzR_2.38.0 Rcpp_1.0.13 Biobase_2.64.0
#> [7] BiocGenerics_0.50.0 ggplot2_3.5.1 massdataset_1.0.34
#> [10] dplyr_1.1.4 magrittr_2.0.3 masstools_1.0.13
#> [13] metid_1.2.32
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 rstudioapi_0.16.0
#> [3] jsonlite_1.8.8 shape_1.4.6.1
#> [5] MultiAssayExperiment_1.30.3 MALDIquant_1.22.3
#> [7] rmarkdown_2.28 GlobalOptions_0.1.2
#> [9] zlibbioc_1.50.0 vctrs_0.6.5
#> [11] RCurl_1.98-1.16 blogdown_1.19
#> [13] progress_1.2.3 htmltools_0.5.8.1
#> [15] S4Arrays_1.4.1 cellranger_1.1.0
#> [17] SparseArray_1.4.8 mzID_1.42.0
#> [19] sass_0.4.9 parallelly_1.38.0
#> [21] bslib_0.8.0 htmlwidgets_1.6.4
#> [23] plyr_1.8.9 impute_1.78.0
#> [25] plotly_4.10.4 cachem_1.1.0
#> [27] igraph_2.0.3 lifecycle_1.0.4
#> [29] iterators_1.0.14 pkgconfig_2.0.3
#> [31] Matrix_1.7-0 R6_2.5.1
#> [33] fastmap_1.2.0 GenomeInfoDbData_1.2.12
#> [35] MatrixGenerics_1.16.0 future_1.34.0
#> [37] clue_0.3-65 digest_0.6.37
#> [39] pcaMethods_1.96.0 colorspace_2.1-1
#> [41] furrr_0.3.1 rprojroot_2.0.4
#> [43] GenomicRanges_1.56.1 fansi_1.0.6
#> [45] httr_1.4.7 abind_1.4-5
#> [47] compiler_4.4.1 here_1.0.1
#> [49] remotes_2.5.0 bit64_4.0.5
#> [51] withr_3.0.1 doParallel_1.0.17
#> [53] BiocParallel_1.38.0 MASS_7.3-61
#> [55] DelayedArray_0.30.1 rjson_0.2.22
#> [57] tools_4.4.1 PSMatch_1.8.0
#> [59] zip_2.3.1 glue_1.7.0
#> [61] QFeatures_1.14.2 grid_4.4.1
#> [63] cluster_2.1.6 reshape2_1.4.4
#> [65] generics_0.1.3 gtable_0.3.5
#> [67] tzdb_0.4.0 preprocessCore_1.66.0
#> [69] tidyr_1.3.1 data.table_1.16.0
#> [71] hms_1.1.3 utf8_1.2.4
#> [73] XVector_0.44.0 foreach_1.5.2
#> [75] pillar_1.9.0 stringr_1.5.1
#> [77] vroom_1.6.5 limma_3.60.4
#> [79] circlize_0.4.16 lattice_0.22-6
#> [81] bit_4.0.5 tidyselect_1.2.1
#> [83] ComplexHeatmap_2.20.0 pbapply_1.7-2
#> [85] knitr_1.48 bookdown_0.40
#> [87] IRanges_2.38.1 SummarizedExperiment_1.34.0
#> [89] xfun_0.47 statmod_1.5.0
#> [91] matrixStats_1.3.0 stringi_1.8.4
#> [93] UCSC.utils_1.0.0 lazyeval_0.2.2
#> [95] yaml_2.3.10 evaluate_0.24.0
#> [97] codetools_0.2-20 MsCoreUtils_1.16.1
#> [99] tibble_3.2.1 BiocManager_1.30.25
#> [101] cli_3.6.3 affyio_1.74.0
#> [103] munsell_0.5.1 jquerylib_0.1.4
#> [105] readxl_1.4.3 GenomeInfoDb_1.40.1
#> [107] globals_0.16.3 png_0.1-8
#> [109] XML_3.99-0.17 parallel_4.4.1
#> [111] readr_2.1.5 prettyunits_1.2.0
#> [113] AnnotationFilter_1.28.0 bitops_1.0-8
#> [115] listenv_0.9.1 viridisLite_0.4.2
#> [117] scales_1.3.0 affy_1.82.0
#> [119] openxlsx_4.2.7 ncdf4_1.23
#> [121] purrr_1.0.2 crayon_1.5.3
#> [123] GetoptLong_1.0.5 rlang_1.1.4
#> [125] vsn_3.72.0