Here, you can learn how to create mass_dataset class object using tidymass.
5.1 Data preparation
The massdataset class object can be used to store the untargeted metabolomics data.
Let’s first prepare the data objects according to the attached figure for each file.
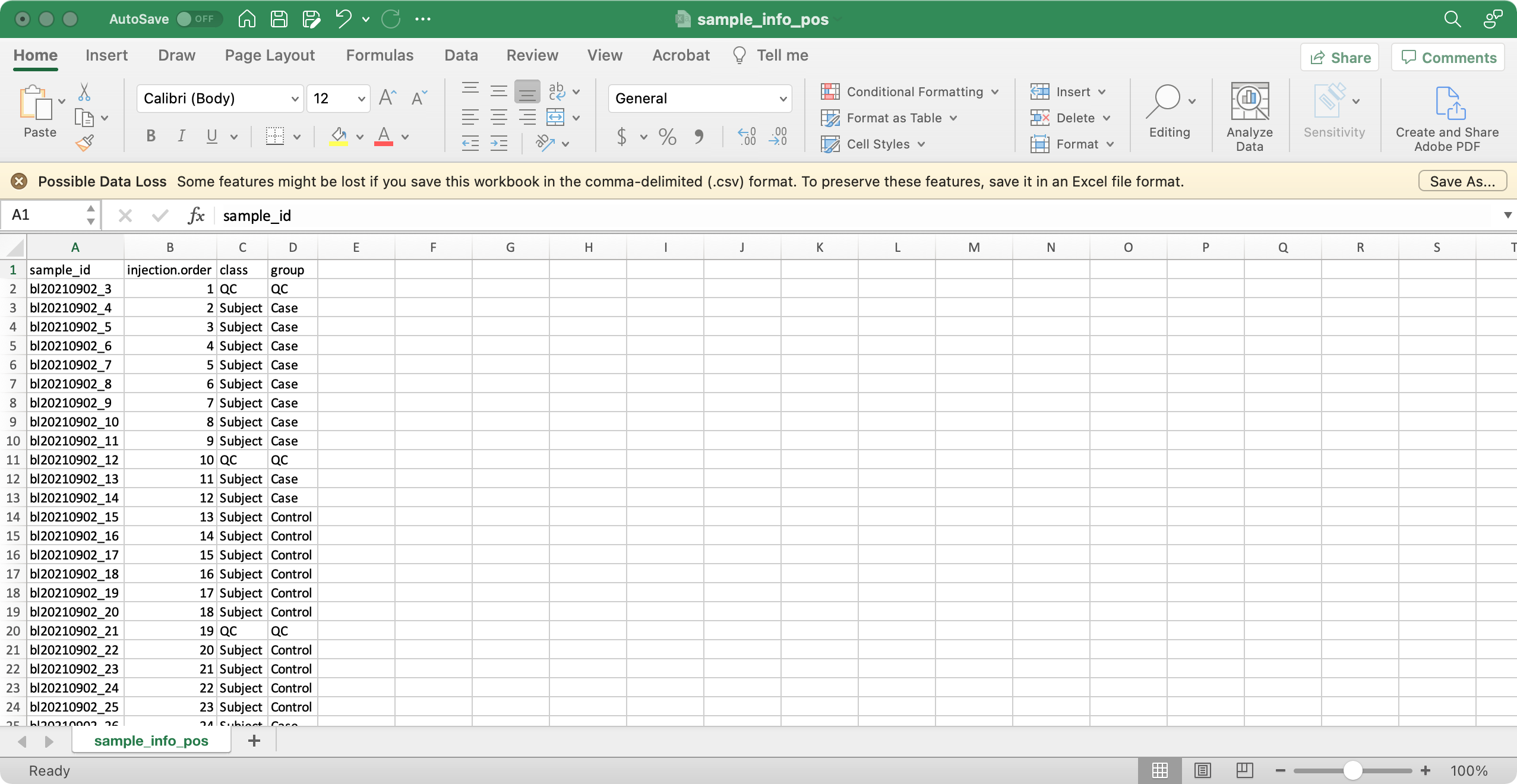
5.1.1sample_info (required)
The columns sample_id (sample ID), injection.order (injection order of samples), class (Blank, QC, Subject, etc), group (case, control, etc) are required.
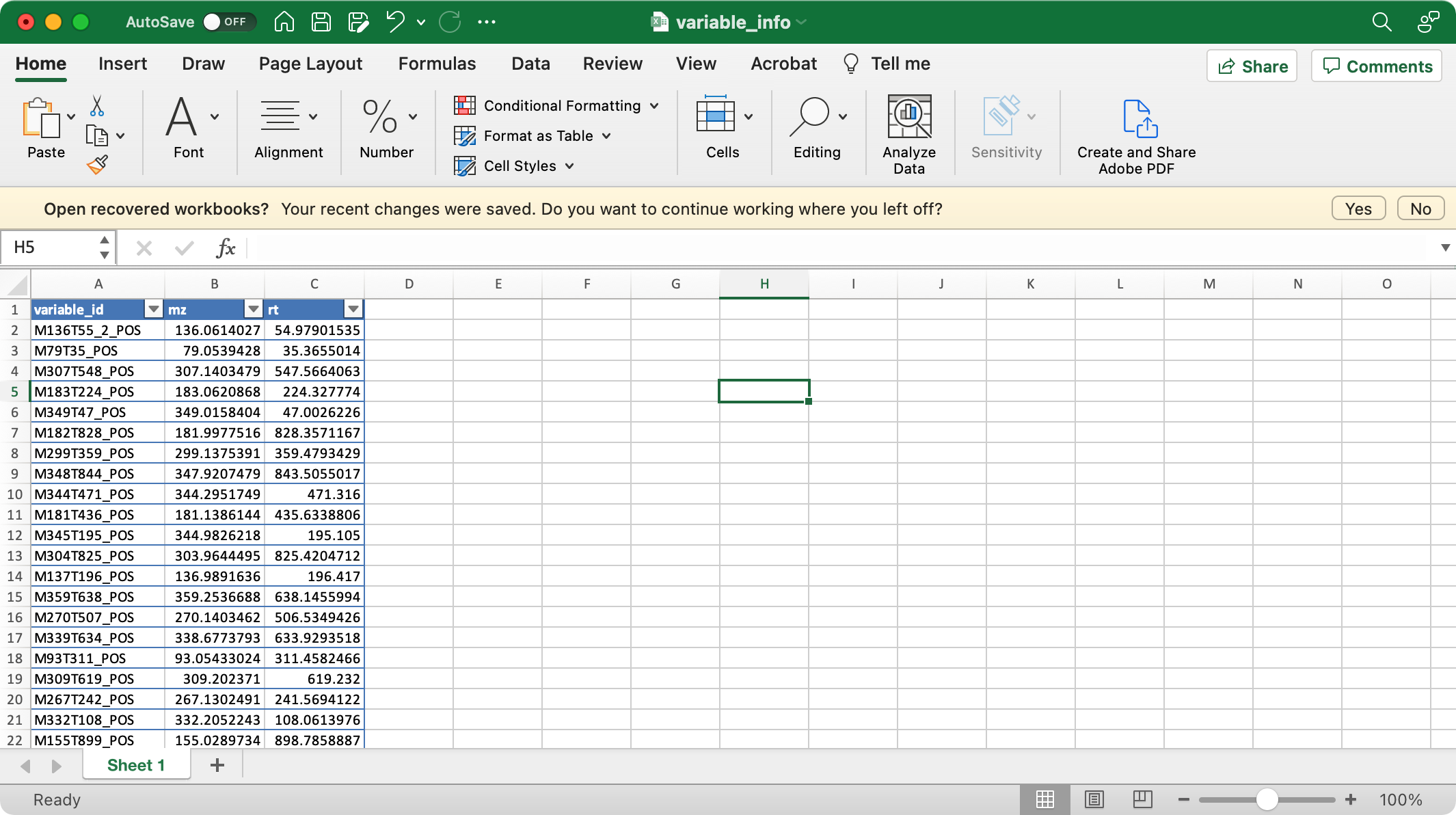
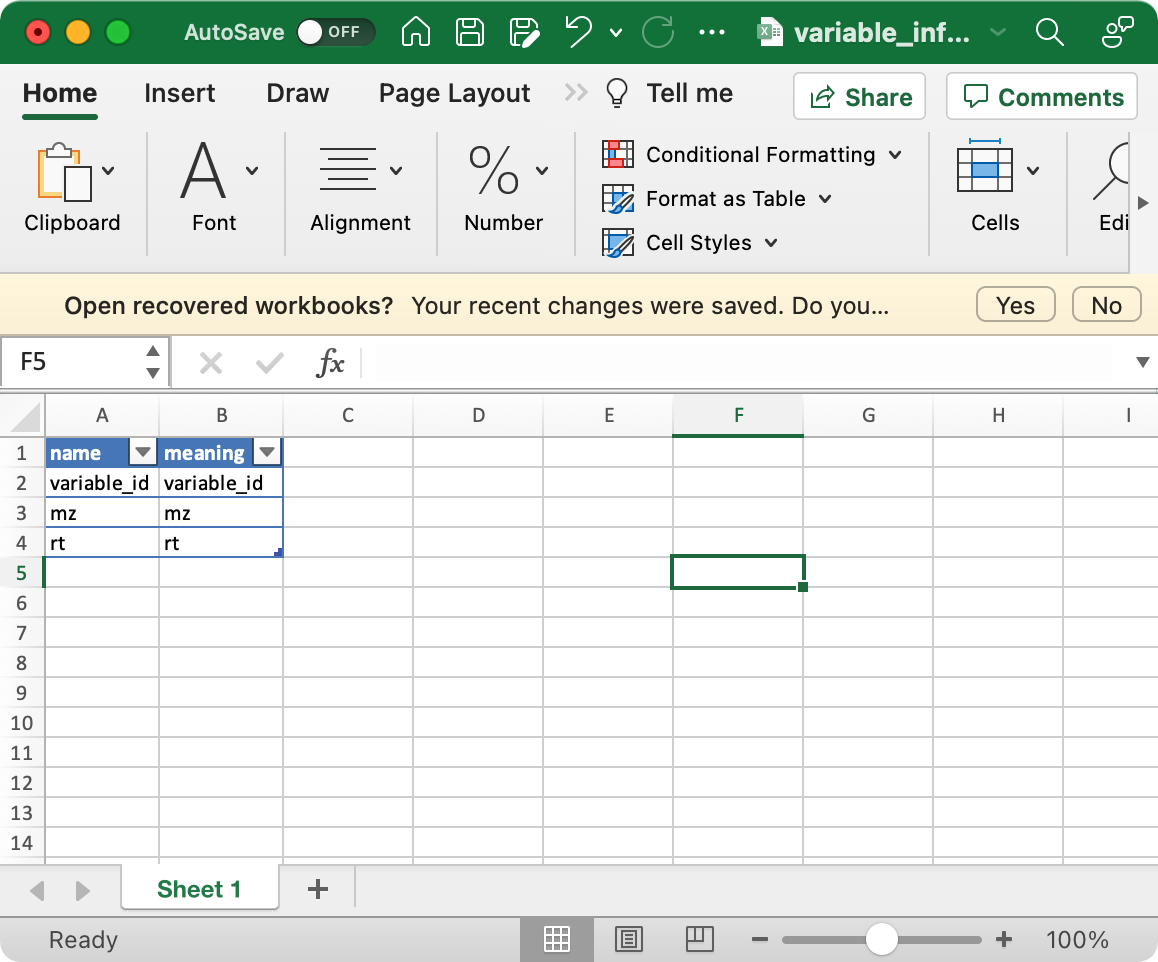
5.1.2variable_info (required)
The columns variable_id (variable ID), mz (mass to charge ratio), rt (retention time, unit is second) are required.
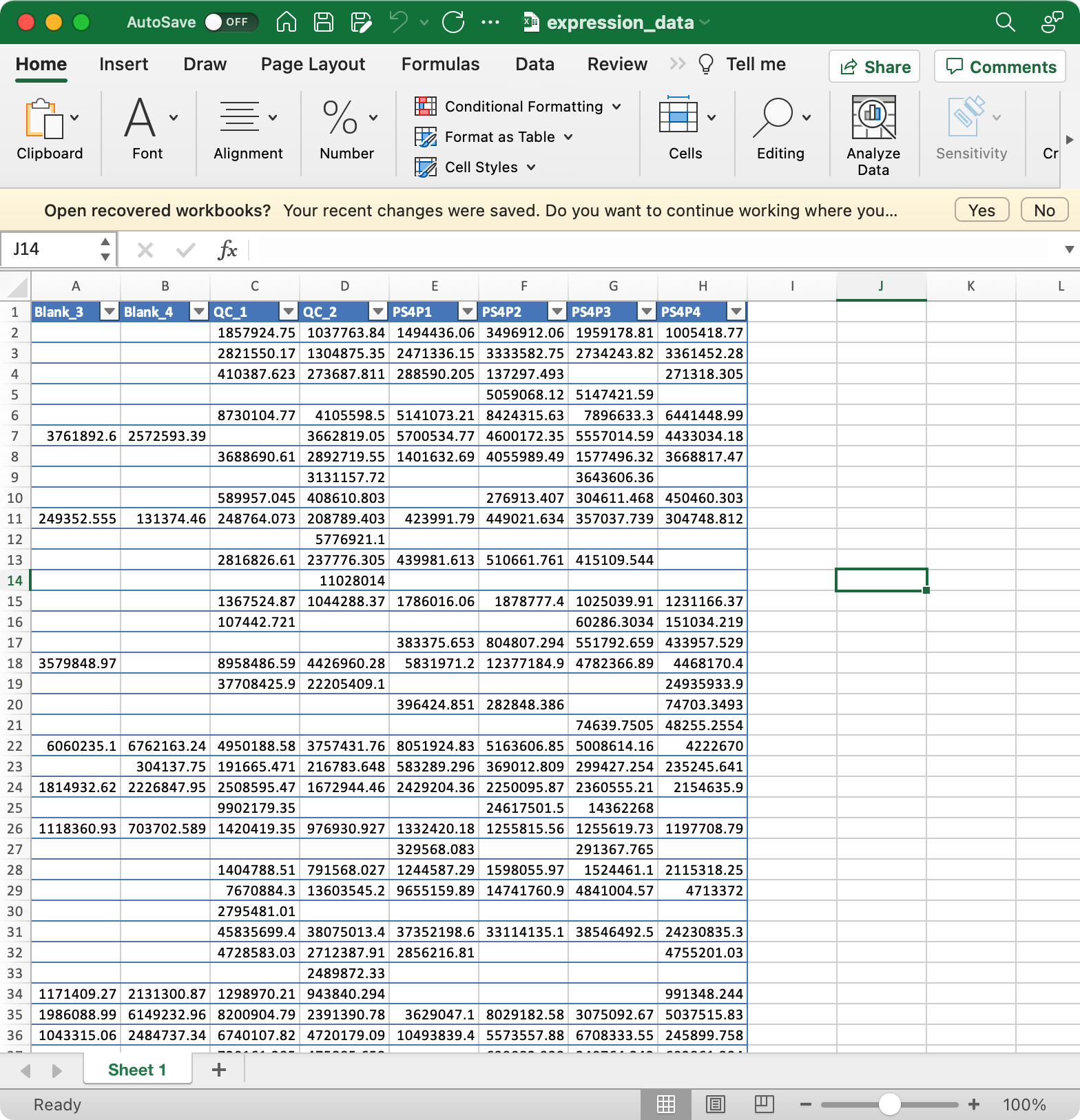
5.1.3expression_data (required)
Columns are samples are rows are features (variables).
The column names of expression_data should be completely same with sample_id in sample_info, and the row names of expression_data should be completely same with variable_id in variable_info.
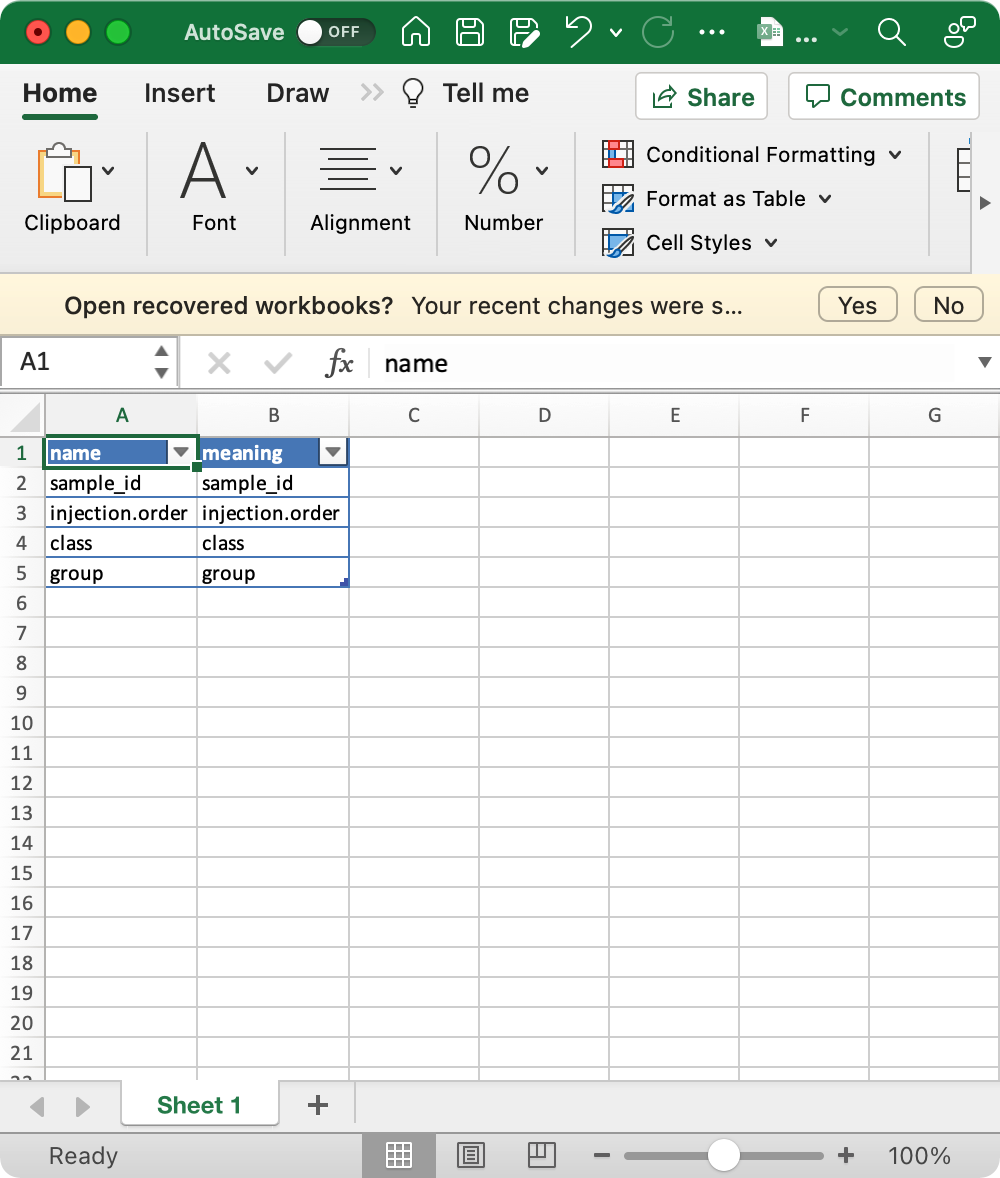
5.1.4sample_info_note (optional)
This is the metadata for sample_info.
5.1.5variable_info_note (optional)
This is the metadata for variable_info.
5.2 Download demo data
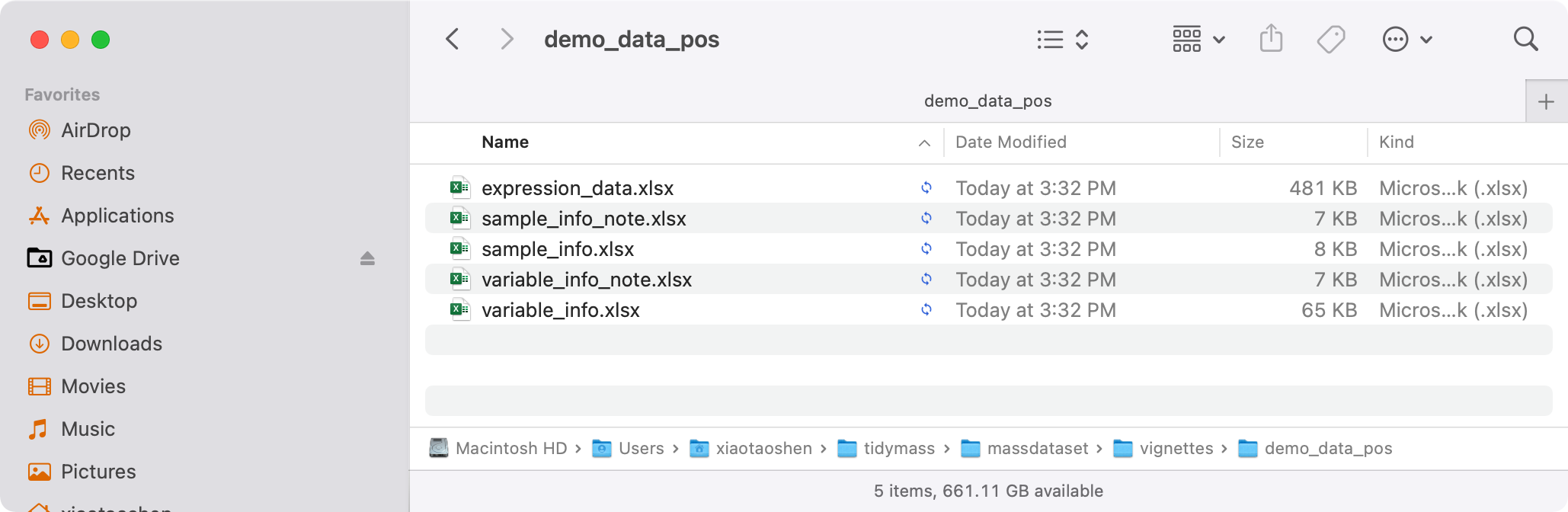
Here we use the demo data from masssprocesser package. The demo data can be downloaded here.
Download this data and uncompress it. And then set the path where you put the folder as working directory.
Then prepare data.
library(tidyverse)
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.1 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
Rows: 1612 Columns: 39
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): variable_id
dbl (38): mz, rt, bl20210902_10, bl20210902_11, bl20210902_13, bl20210902_14...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 5486 Columns: 39
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): variable_id
dbl (38): mz, rt, X20210902_neg04, X20210902_neg05, X20210902_neg06, X202109...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 36 Columns: 4
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (3): sample_id, class, group
dbl (1): injection.order
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 36 Columns: 4
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (3): sample_id, class, group
dbl (1): injection.order
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Variable information and expression data are in the peak table. Let’s separate them.
 ## Session information
## Session information